5. 基本機能を利用した計算例
5.1. 全エネルギー計算
最も基本的な計算機能として、全エネルギーの計算があります。複数の格子定数で全エネルギーを計算し格子定数や体積弾性率を計算することや、絶対零度における結晶の安定性を評価することができます。
5.1.1. 入力パラメータ
シリコン結晶(ダイヤモンド構造)の全エネルギー計算を例とします。この例題の入力ファイルは samples/basic/Si8 以下にあります。
シリコン原子8個の系Si8を対象とします。シリコン結晶(ダイヤモンド構造)の構造を 図 5.1 に示します。

図 5.1 シリコン原子が構成するダイヤモンド構造
計算に使う入力ファイルは、ファイルfile_names.dataの中で指定します。
file_names.dataは以下のように記述します。
&fnames
F_INP = './input_scf_Si8.data'
F_POT(1) = '../../pp/Si_ggapbe_paw_nc_01m.pp'
...
F_CHR = './nfchr.cube'
/
PHASEを実行するためには、擬ポテンシャルデータ F_POT(1) と、入力ファイルF_INPが指定されている必要があります。 Si_ggapbe_paw_nc_01m.pp はシリコンの擬ポテンシャル・データです。
入力パラメータファイルinput_scf_Si8.data について説明します。
Controlブロックでは、全体的な計算条件を指定します。cpumaxは計算時間の最大値を指定しています。
Control{
condition = initial
cpumax = 3600 sec ! {sec|min|hour|day}
}
Accuracyブロックでは、計算精度を指定します。
accuracy{
cutoff_wf = 20.00 rydberg
cutoff_cd = 80.00 rydberg
num_bands = 20
ksampling{
method = mesh ! {mesh|file|directin|gamma}
mesh{ nx = 4, ny = 4, nz = 4 }
}
...
scf_convergence{
delta_total_energy = 1.e-10 hartree
succession = 2
}
...
}
cutoff_wf と cutoff_cd は、 波動関数と電荷密度分布のカットオフ・エネルギーが、 それぞれ 20.0 Ry と80.0 Ry という値であることを表しています。
num_bands はエネルギー準位数を表します。この計算では、Si原子8個を扱いますが、各原子は4個の価電子をもつため、占有される準位数は、スピンの縮退度を考慮すると8\(\times\)4/2=16となります。 このため num_bands は、17以上に設定しておく必要があります。 また、ksampling というタグは、\(k\)点のサンプリングの方法を 指定するのに使われます。この例では、\(4 \times 4 \times 4\)のメッシュ点がk点サンプリングとなります。
scf_convergenceでは、計算の収束条件を指定します。この例の場合、全エネルギーの計算誤差が\(10^{- 10}\) Hartree 未満に収まるという結果が連続して2回続いたら、計算を終了させるように指定されています。
Structureブロックでは、結晶構造を指定します。単位はデフォルトが原子単位となっています(長さの単位は Bohr)。
structure{
unit_cell_type = primitive
unit_cell{
a_vector = 10.26 0.00 0.00
b_vector = 0.00 10.26 0.00
c_vector = 0.00 0.00 10.26
}
atom_list{
coordinate_system = internal ! {cartesian|internal}
atoms{
#default weight = 1, element = Si, mobile = 1
#tag rx ry rz
0.125 0.125 0.125
-0.125 -0.125 -0.125
0.125 0.625 0.625
-0.125 -0.625 -0.625
0.625 0.125 0.625
-0.625 -0.125 -0.625
0.625 0.625 0.125
-0.625 -0.625 -0.125
}
}
element_list{ #tag element atomicnumber
Si 14
}
}
atom_list では、原子種、単位胞内での内部座標位置、それぞれの原子の位置を固定するか否かを指定します。 element_list では、元素名とその原子番号を指定します。
Postprocessingブロックでは、後処理のパラメータを指定します。
postprocessing{
...
charge{
sw_charge_rspace = ON
filetype = cube !{cube|density_only}
title = "This is a title line for the bulk Si"
}
}
chargeブロックでは、電荷密度の出力について指定します。電荷密度は、file_names.dataにおいて F_CHR で指定したファイルに出力されます。 filetype = cube とする事により、Gaussian cube 形式で出力されます。 このとき、F_CHRで指定されるファイル名は、*.cube の形式である必要があります。 Gaussian cubeファイルは、PHASE Viewerなどの可視化ソフトウェアを使って可視化表示することが可能です。
5.1.2. 計算の実行
PHASEを以下のように実行します。
% mpirun -np NP ../../../bin/phase ne=NE nk=NK
ここで、NP、NE、NK はそれぞれ、計算に使用するプロセッサーの数、 エネルギー準位の分割計算の数、および、\(k\)点の分割計算の数を指します。これらのパラメータの値の間には、NP = NE×NK という 関係が成り立っていなければなりません。
また、1 CPU の計算機を使う場合には、 以下のように実行します。
% mpirun ../../../bin/phase
計算の途中経過を確認するには、計算のログ出力ファイルoutput000 に出力されている全エネルギーの収束状況を調べます。以下のように実行すると、全エネルギーに関する部分を抽出できます。
% grep TH output000
Si8 のサンプルを使って得られた output000では、次のような結果が表示されます。
TOTAL ENERGY FOR 1 -TH ITER= -30.525762143533 EDEL = -0.305258D+02 : SOLVER = MATDIAGON : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 2 -TH ITER= -31.439227176416 EDEL = -0.913465D+00 : SOLVER = SUBMAT + PKOSUGI : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 3 -TH ITER= -31.469956871528 EDEL = -0.307297D-01 : SOLVER = SUBMAT + PKOSUGI : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 4 -TH ITER= -31.487810280728 EDEL = -0.178534D-01 : SOLVER = SUBMAT + PKOSUGI : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 5 -TH ITER= -31.495578938717 EDEL = -0.776866D-02 : SOLVER = SUBMAT + PKOSUGI : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 6 -TH ITER= -31.500918535399 EDEL = -0.533960D-02 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 7 -TH ITER= -31.501113667547 EDEL = -0.195132D-03 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 8 -TH ITER= -31.501186121230 EDEL = -0.724537D-04 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 9 -TH ITER= -31.501187563396 EDEL = -0.144217D-05 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 10 -TH ITER= -31.501187881041 EDEL = -0.317645D-06 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 11 -TH ITER= -31.501187967072 EDEL = -0.860305D-07 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 12 -TH ITER= -31.501187974714 EDEL = -0.764159D-08 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 13 -TH ITER= -31.501187977198 EDEL = -0.248405D-08 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 14 -TH ITER= -31.501187977591 EDEL = -0.393619D-09 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
TOTAL ENERGY FOR 15 -TH ITER= -31.501187977742 EDEL = -0.150855D-09 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = BROYD2
SCF計算において、全エネルギーの値が収束してゆく様子が分かります。
5.1.3. 計算結果の出力
計算された全エネルギーは、F_ENF ファイルに出力されます。
Si8の例題では、 F_ENF ファイル(ファイル名:nfefn.data)は以下のようになっています。
iter_ion, iter_total, etotal, forcmx
1 15 -31.5087632805 0.0000104146
計算が終了すると、電荷密度ファイルnfchr.cubeが作成されます。電荷密度分布を 図 5.2 に示します。原子数を増やすなど、cube file に修正を加えています。

図 5.2 シリコン結晶の電荷密度分布
5.2. 対称性を考慮した計算
PHASEには、結晶の対称性を考慮することによって計算量を低減する機能があります。対称性は、自動的に判定させることも可能ですし、生成元を直接指定する ことによって明示的に指定することも可能です。 原子配置を指定する方法には, 基本格子を指定する方法とブラベー格子を指定する方法があります。 具体的には, 変数 unit_cell_type の入力値を primitive か Bravais のどちらかから選択することで 指定できます。
5.2.1. 入力パラメータ
5.2.1.1. 単位胞の指定
単位胞を基本格子で指定
unit_cell_type = primitive
unit_cell{
#units bohr
a_vector = 0.00000 5.13000 5.13000
b_vector = 5.13000 0.00000 5.13000
c_vector = 5.13000 5.13000 0.00000
}
この方法は, unit_cell_type がprimitive でもBravais でも使用できます。
単位胞を格子定数で指定
unit_cell_type = Bravais
unit_cell{
#units bohr
a = 10.26, b = 10.26, c = 10.26
alpha = 90, beta = 90, gamma = 90
}
この方法は, unit_cell_type がBravais の時のみ使用できます。ブラベー格子を指定して入力した場合、対称性の指定により, プログラム内で基本格子を決定します。計算は、プログラムが決定した基本格子を元に行われるので、原子座標の指定、k点分割数や、バンド計算時の対称k点の指定などは、この基本格子を元に行う必要がある点に注意ください。
unit_cell_type としてBravais を利用する場合、副格子点に位置する原子は指定しないようにしてください。たとえば体心原子を含む結晶の場合, (0, 0, 0) の原子は指定し, (0.5, 0.5, 0.5) の原子は指定しないようにしてください。Bravais を利用する際に指定が必要な結晶の型は、tspaceブロックの下のlattice_system という変数で指定します。具体的には以下のように指定します。
structure{
unit_cell_type = Bravais
tspace{
lattice_system = facecentered
}
}
lattice_systemにおいて指定できるパラメーターについては表 5.1 を参照してください。
菱面体晶系(rhombohedral) の場合には, 対応する六方晶系(hexagonal) の格子定数を入力します。六方晶系と菱面体晶系の基本並進ベクトルの関係を 図 5.3 に示します.。
原子座標を内部座標で入力する場合は, 等価原子を除いて, 単位胞内のすべて原子の位置を結晶軸ベクトル(慣用単位胞の3 辺を表すベクトル) に対する相対座標(ワイコフ位置の原子座標) で入力します。デカルト座標で入力する場合は, 表 5.2 に示されている基本並進ベクトルと整合するように入力してください。
晶系 |
格子定数 |
unit_cellに 記述する値 |
格子の種類 |
lattic _systemに指 定する単語 |
|---|---|---|---|---|
立方(c) |
\(a\) |
a= \(a\), b= \(a\), c= \(a\) alpha=90, beta=90, gamma=90 |
単純(P) 面心(F) 体心(I) |
primitive facecentered bodycentered |
正方(t) |
\(a,c\) |
a= \(a\), b= \(a\), c= \(c\) alpha=90, beta=90, gamma=90 |
単純(P) 体心(I) |
primitive bodycentered |
直方(o) |
\(a,b\) \(,c\) |
a= \(a\), b= \(b\), c= \(c\) alpha=90, beta=90, gamma=90 |
単純(P) 底心(C) 面心(F) 体心(I) |
primitive basecentered facecentered bodycentered |
六方(h) |
\(a,c\) |
a= \(a\), b= \(a\), c= \(c\) alpha=90, beta=90, gamma=120 |
単純(P) |
hexagonal |
三方(h) 菱面体軸 六方晶軸 |
\(a,c\) |
a= \(a\), b= \(a\), c= \(c\) alpha=90, beta=90, gamma=120 |
菱面(R) 単純(P) |
rhombohedral hexagonal |
単斜(m) |
\(a,b\) \(,c\) \(\beta\) |
a= \(a\), b= \(b\), c= \(c\) alpha=90, beta= \(\beta\) gamma=90 |
単純(P) 底心(C) |
primitive basecentered |
三斜(a) |
\(a,b,c\) \(\alpha,\beta\) \(\gamma\) |
a= \(a\), b= \(b\), c= \(c\) alpha=\(\alpha\) beta=\(\beta\) gamma=\(\gamma\) |
単純(P) |
primitive |

図 5.3 六方晶系と菱面体晶系の関係. 六方軸の方から見た格子点と基本並進ベクトルが示されています. \(\mathbf{a}^{\mathbf{H}}\mathbf{,}\mathbf{b}^{\mathbf{H}}\mathbf{,}\mathbf{c}^{\mathbf{H}}\)は六方晶系の基本並進ベクトルで, \(\mathbf{a}^{\mathbf{R}}\mathbf{,}\mathbf{b}^{\mathbf{R}}\mathbf{,}\mathbf{c}^{\mathbf{R}}\)は菱面体晶系の基本並進ベクトルです.
ブラベー格子 |
\(\mathbf{a}\) |
\(\mathbf{b}\) |
\(\mathbf{c}\) |
単純立方(cP) |
\(a\widehat{\mathbf{x}}\) |
\(a\widehat{\mathbf{y}}\) |
\(a\widehat{\mathbf{z}}\) |
面心立方(cF) |
\(\frac{a}{2}(\widehat{\mathbf{y}} + \widehat{\mathbf{z}})\) |
\(\frac{a}{2} (\widehat{\mathbf{x}}+\widehat{\mathbf{y}} )\) |
\(\frac{a}{2}(\widehat{\mathbf{x}}+\widehat{\mathbf{y}}\) |
体心立方(cI) |
\(\frac{a}{2} (-\widehat{\mathbf{x}} + \widehat{\mathbf{y}} + \widehat{\mathbf{z}})\) |
\(\frac{a}{2} (\widehat{\mathbf{x}} - \widehat{\mathbf{y}} + \widehat{\mathbf{z}})\) |
\(\frac{a}{2} (\widehat{\mathbf{x}} + \widehat{\mathbf{y}} - \widehat{\mathbf{z}})\) |
単純正方(tP) |
\(a \widehat{\mathbf{x}}\) |
\(a \widehat{\mathbf{y}}\) |
\(c \widehat{\mathbf{z}}\) |
体心正方(tI) |
\(\frac{1}{2} ( -a \widehat{\mathbf{x}} + a \widehat{\mathbf{y}} + c \widehat{\mathbf{z}})\) |
\(\frac{1}{2}(a \widehat{\mathbf{x}} - b \widehat{\mathbf{y}} + c \widehat{\mathbf{z}})\) |
\(\frac{1}{2} (a \widehat{\mathbf{x}} + a \widehat{\mathbf{y}} - c \widehat{\mathbf{z}})\) |
単純直方(oP) |
\(a \widehat{\mathbf{x}}\) |
\(b \widehat{\mathbf{y}}\) |
\(c \widehat{\mathbf{z}}\) |
底心直方(oC) |
\(\frac{1}{2}(a \widehat{\mathbf{x}}-b\widehat{\mathbf{y}})\) |
\(\frac{1}{2}(a\widehat{\mathbf{x}}+b\widehat{\mathbf{y}})\) |
\(c\widehat{\mathbf{z}}\) |
面心直方(oF) |
\(\frac{1}{2}(b\widehat{\mathbf{y}}+c\widehat{\mathbf{z}})\) |
\(\frac{1}{2}(a\widehat{\mathbf{x}}+c\widehat{\mathbf{z}})\) |
\(\frac{1}{2}(a\widehat{\mathbf{x}}+c\widehat{\mathbf{y}})\) |
体心直方(oI) |
\(\frac{1}{2}(-a\widehat{\mathbf{x}}+b\widehat{\mathbf{y}}+c\widehat{\mathbf{z}})\) |
\(\frac{1}{2}(a\widehat{\mathbf{x}}-b\widehat{\mathbf{y}}+c\widehat{\mathbf{z}})\) |
\(\frac{1}{2}(a\widehat{\mathbf{x}}+b\widehat{\mathbf{y}}-c\widehat{\mathbf{z}})\) |
単純六方(hP) |
\(a\widehat{\mathbf{x}}\) |
\(a(-\frac{1}{2}\widehat{\mathbf{x}}+\frac{\sqrt{3}}{2}\widehat{\mathbf{y}})\) |
\(c\widehat{\mathbf{z}}\) |
単純菱面体(hR) |
\(\frac{a}{2}\widehat{\mathbf{x}}+\frac{a}{2\sqrt{3}}\widehat{\mathbf{y}}+\frac{1}{3}c\widehat{\mathbf{z}}\) |
\(-\frac{a}{2}\widehat{\mathbf{x}}+\frac{a}{2\sqrt{3}}\widehat{\mathbf{y}}+\frac{1}{3}c\widehat{\mathbf{z}}\) |
\(-\frac{a}{\sqrt{3}}\widehat{\mathbf{y}} + \frac{1}{3} c \widehat{\mathbf{z}}\) |
単純単斜(mP) |
\(a\widehat{\mathbf{x}}\) |
\(b\widehat{\mathbf{y}}\) |
\(c(\cos \beta \widehat{\mathbf{x}} + \sin \beta \widehat{\mathbf{z}})\) |
底心単斜(mC) |
\(\frac{1}{2}(a \widehat{\mathbf{x}} - b \widehat{\mathbf{y}})\) |
\(\frac{1}{2}(a \widehat{\mathbf{x}}+b\widehat{\mathbf{y}})\) |
\(c(\cos \beta \widehat{\mathbf{x}} + \sin \beta \widehat{\mathbf{z}})\) |
単純三斜(aP) |
\(a\widehat{\mathbf{x}}\) |
\(b( \cos \gamma \widehat{\mathbf{x}} + \sin \gamma \widehat{\mathbf{y}})\) |
\(c\left( \cos \beta \widehat{\mathbf{x}} + \frac{\cos \alpha - \cos\beta \cos\gamma}{\sin \gamma} \widehat{\mathbf{y}}\right)\) \(+ \sqrt{1-\frac{\cos^2\alpha + \cos^2\beta-2\cos\alpha\cos\beta\cos\gamma}{sin^2\gamma}} \widehat{\mathbf{z}}\) |
5.2.1.2. 対称性の指定
対称性の指定のやり方には, 結晶構造を入力する方法、対称操作を自動的に決定する方法、生成元を入力する方法があります.
結晶構造を入力する方法
変数 crystal_structure に, 結晶構造の型を入力します. この場合, 選択肢には diamond, hexagonal, fcc, bcc, simple_cubic の5つがあります。 Si結晶の場合に指定する結晶構造は diamond です.
対称操作を自動的に決定する方法
method変数にautomaticを指定することで, 対称性は自動的に決定されます。 tspaceブロックのlattice_systemに格子の型を指定します。 省略すると既定値のprimitiveが採用されます。
symmetry{
method = automatic
tspace{
lattice_system = facecentered !{rhombohedral|hexagonal|primitive|facecentered|bodycentered|basecentered}
}
}
生成元を入力する方法
生成元は、tspace ブロックで指定します。Si結晶の場合, tspace の入力値は以下のようになります.
tspace{
lattice_system = facecentered !{rhombohedral|hexagonal|primitive|facecentered|bodycentered|basecentered}
num_generators = 3
generators{
#tag rotation tx ty tz
IE 0 0 0
C31+ 0 0 0
C4X+ 1/4 1/2 3/4
}
}
予め, 面心格子を使うことを lattice_system = facecentered で, また, 生成元の数が3であることを num_generators = 3 で宣言した後で, タグ generators の中で, IE, C31+, C4X+ が, 具体的に3種類の生成元を指定しています。
生成元の指定の方法を説明します。
生成元の回転操作は, 以下のコードで指定します. 各行は, それぞれ一つの回転操作に対応します. 一列目の数字か二列目の記号を利用してgeneratorsテーブルのrotation列に対称操作を指定します. 三列目から五列目までが対応する回転操作を表します. なお, 三方晶, 六方晶の場合に現れているWはX-Yを表します. コードは, 一列目の数字でも二列目の文字列でも指定することが可能です.
三方晶, 六方晶の場合
1 E X Y Z 13 IE -X -Y -Z
2 C6+ W X Z 14 IC6+ -W -X -Z
3 C3+ -Y W Z 15 IC3+ Y -W -Z
4 C2 -X -Y Z 16 IC2 X Y -Z
5 C3- -W -X Z 17 IC3- W X -Z
6 C6- Y -W Z 18 IC6- -Y W -Z
7 C211 -W Y -Z 19 IC211 W -Y Z
8 C221 X W -Z 20 IC221 -X -W Z
9 C231 -Y -X -Z 21 IC231 Y X Z
10 C212 W -Y -Z 22 IC212 -W Y Z
11 C222 -X -W -Z 23 IC222 X W Z
12 C232 Y X -Z 24 IC232 -Y -X Z
三方晶, 六方晶以外の場合
1 E X Y Z 25 IE -X -Y -Z
2 C2X X -Y -Z 26 IC2X -X Y Z
3 C2Y -X Y -Z 27 IC2Y X -Y Z
4 C2Z -X -Y Z 28 IC2Z X Y -Z
5 C31+ Z X Y 29 IC31+ -Z -X -Y
6 C32+ -Z X -Y 30 IC32+ Z -X Y
7 C33+ -Z -X Y 31 IC33+ Z X -Y
8 C34+ Z -X -Y 32 IC34+ -Z X Y
9 C31- Y Z X 33 IC31- -Y -Z -X
10 C32- Y -Z -X 34 IC32- -Y Z X
11 C33- -Y Z -X 35 IC33- Y -Z X
12 C34- -Y -Z X 36 IC34- Y Z -X
13 C2A Y X -Z 37 IC2A -Y -X Z
14 C2B -Y -X -Z 38 IC2B Y X Z
15 C2C Z -Y X 39 IC2C -Z Y -X
16 C2D -X Z Y 40 IC2D X -Z -Y
17 C2E -Z -Y -X 41 IC2E Z Y X
18 C2F -X -Z -Y 42 IC2F X Z Y
19 C4X+ X -Z Y 43 IC4X+ -X Z -Y
20 C4Y+ Z Y -X 44 IC4Y+ -Z -Y X
21 C4Z+ -Y X Z 45 IC4Z+ Y -X -Z
22 C4X- X Z -Y 46 IC4X- -X -Z Y
23 C4Y- -Z Y X 47 IC4Y- Z -Y -X
24 C4Z- Y -X Z 48 IC4Z- -Y X -Z
他方, 回転に伴う並進操作はgeneratorsテーブルのtx, ty, tz列にそれぞれ指定します. 格子ベクトルを基準に分数で入力してください.
5.2.1.3. 反転対称性がある場合
反転対称がある場合, これを考慮する事により, 計算量を減らすことができます。 たとえば、以下の座標データは原点を中心として反転対称性があるので、それを考慮するように設定すると計算量を減らすことができます。
atom_list{
coordinate_system = internal ! {cartesian|internal}
atoms{
#units !{angstrom(cartesian) bohr(cartesian)}
#tag rx ry rz weight element mobile
0.125 0.125 0.125 1 Si 1
-0.125 -0.125 -0.125 1 Si 1
}
}
反転対称性を考慮する設定は、symmetryブロックの下でsw_inversion = onとすることによって行います。
structure{
...
symmetry{
...
sw_inversion = on
}
}
また、反転対称性を考慮する場合、原子配置のweight属性値を利用することによって座標データ入力を省力化することも可能です。たとえば、以下の指定はsw_inversion=onの場合上記の座標例と等価です。
atom_list{
coordinate_system = internal ! {cartesian|internal}
atoms{
#units !{angstrom(cartesian) | bohr(cartesian)}
#tag rx ry rz weight element mobile
0.125 0.125 0.125 2 Si 1
}
symmetry{
sw_inversion = on
}
}
weight属性値が2の原子は、反転対称位置に自分自身のコピーが作成されます。
前節で指定した対称群に反転対称操作が含まれる場合、このoptionを指定することを推奨します。 なお、原子座標を入力する場合反転対称操作の中心は原点であることにご注意ください。また、反転対称性のない系においてsw_inversion = onを指定するとエラーメッセージを出力して計算を終了します。
5.2.2. 計算例:シリコン結晶(Si2)
シリコン原子が構成するダイヤモンド構造の基本格子は原子2個を含みます。ここでは、シリコン原子2個からなる Si2という系を例とします。 図 5.4 はSi2の原子構造です。
計算例題は、 samples/basic/Si2 です。

図 5.4 Si2の原子構造。黄線は原子2個を含む基本格子を表す
SCF計算
SCF計算を行い、電荷密度を計算します。計算例題は samples/basic/Si2/scf です。
ファイル file_names.data において、入力パラメータファイルと擬ポテンシャルを指定します。
F_POT(1) = '../../pp/Si_ggapbe_paw_nc_01m.pp'
F_CHGT = '../scf/nfchgt.data'
...
入力パラメータファイルにおいて、crystal_structure をdiamondとして、対称性を指定します。
accuracy{
cutoff_wf = 20.00 rydberg
cutoff_cd = 80.00 rydberg
num_bands = 8
}
structure{
unit_cell_type = Bravais
unit_cell{
a = 10.26, b = 10.26, c = 10.26
alpha = 90, beta = 90, gamma = 90
}
symmetry{
crystal_structure = diamond
}
atom_list{
atoms{
#tag rx ry rz element
0.125 0.125 0.125 Si
-0.125 -0.125 -0.125 Si
}
}
}
エネルギー準位数を表す num_bands の値は、原子数が2個なので8としています。
PHASEを実行します。
% mpirun ../../../../bin/phase
計算が終了すると、file_names.data というファイルの中で、 変数 F_CHGT で指定した出力ファイル nfchgt.data に、 計算によって得られた電荷の情報が出力されます。
状態密度(DOS)の計算
状態密度(DOS)を計算します。計算例題は、 samples/basic/Si2/dos です。
計算結果の出力ファイルが上書きされるのを避けるため、SCF計算を行ったディレクトリとは別のディレクトリで実行します。
SCF計算結果の電荷密度ファイルnfchgt.data を使います。擬ポテンシャルはSCF計算と同じものを使います。 file_names.data では、入出力ファイルを以下のように指定しています。
F_INP = './input_dos_Si.data'
F_POT(1) = '../../pp/Si_ggapbe_paw_nc_01m.pp'
...
F_CHGT = '../scf/nfchgt.data'
... ...
F_ENERG = './nfenergy.data'
... ...
F_CHGTで指定している電荷密度のデータは、SCF計算で得られた出力ファイルです。 入力ファイルは input_dos_Si.data と nfchgt.data の2つです。 入力ファイル input_dos_Si.data について、SCF計算の入力ファイル input_scf_Si.data と異なる部分を以下に示します。
Control{
condition = fixed_charge
}
accuracy{
cutoff_wf = 20.00 rydberg
cutoff_cd = 80.00 rydberg
num_bands = 8
ksampling{
method = mesh
mesh{ nx = 4, ny = 4, nz = 4 }
}
smearing{
method = tetrahedral
}
initial_wavefunctions = matrix_diagon
matrix_diagon{
cutoff_wf = 9.00 rydberg
}
ek_convergence{
num_max_iteration = 200
sw_eval_eig_diff = on
delta_eigenvalue = 1.e-8 hartree
succession = 2
}
}
postprocessing{
dos{
sw_dos = ON
method = tetrahedral !{ tetrahedral | Gaussian }
deltaE_dos = 1.e-3 eV
nwd_window_width = 10
}
}
Controlブロックのconditionをfixed_chargeと設定することによってSCF計算で得られた電荷の分布を固定して使用することを指定します。ksamplingでは\(k\)点サンプリングが\(4 \times 4 \times 4\)であることを指定しています。smearingでは四面体法を用いることを指定しています。ek_convergenceブロックでは固定電荷計算の収束条件を指定しています。 Postprocessingブロックでは、計算終了後の後処理として、四面体法による状態密度の計算のパラメータが指定されています。 これらの入力ファイルを使って、プログラムekcalを用いて、状態密度の計算を行います。
% mpirun ../../../../bin/ekcal
計算を実行すると、nfenergy.data という出力ファイルが生成されます。 これは、各\(k\)点ごとのエネルギー値を、エネルギーの低い方から順に 出力したもので、その最初の部分は以下のようになっています。
num_kpoints = 141
num_bands = 8
nspin = 1
Valence band max = 0.233846
=== energy_eigen_values ===
ik = 1 ( 0.000000 0.500000 0.500000 )
-0.0484324491 -0.0484324491 0.1258095002 0.1258095002
0.2619554320 0.2619554320 0.6015285289 0.6015285289
=== energy_eigen_values ===
ik = 2 ( 0.000000 0.490000 0.490000 )
-0.0540717117 -0.0427149546 0.1258687813 0.1258687813
0.2607026827 0.2633829946 0.6006244013 0.6006244013
=== energy_eigen_values ===
ik = 3 ( 0.000000 0.480000 0.480000 )
-0.0596299923 -0.0369220783 0.1260465996 0.1260465996
0.2596226501 0.2649874134 0.5980547648 0.5980547648
=== energy_eigen_values ===
ik = 4 ( 0.000000 0.470000 0.470000 )
-0.0651046420 -0.0310567694 0.1263428799 0.1263428799
0.2587131916 0.2667706685 0.5941566835 0.5941566835
=== energy_eigen_values ===
ik = 5 ( 0.000000 0.460000 0.460000 )
-0.0704931128 -0.0251220735 0.1267574962 0.1267574962
0.2579721226 0.2687346642 0.5892968047 0.5892968047
最初の2行は、それぞれ、\(k\)点とバンドの数を表します。 3行目は、この計算でスピン分極は考慮されていないことを、 また、4行目は価電子帯上端におけるエネルギーの値を指しています。
ツールdos.pl を使って、 電子状態密度の図を作成します。 描画するエネルギー範囲の最小値 E1 と最大値 E2 を決めて、
% dos.pl dos.data -erange=E1,E2 -color
とすると、Postscript 形式の状態密度図 density_of_states.eps が得られます。 また、-with_fermi というオプションをつけて、この処理を実行すると、生成される 状態密度図にフェルミ・レベルが点線で描かれます。 ただし、ギャップのある系では、 価電子帯のエネルギー最大値のところに点線が引かれます。
この例題では、以下のように実行します。
% dos.pl dos.data -erange=-13,5 -with_fermi -color
Si2の状態密度を、図 5.5 に示します。

図 5.5 Si2の状態密度
バンド構造図
バンド構造を計算します。計算例題は、 samples/basic/Si2/band です。
file_names.data では、入出力ファイルを以下のように指定しています。
F_INP = './input_band_Si.data'
F_POT(1) = '../../pp/Si_ggapbe_paw_nc_01m.pp'
F_KPOINT = './kpoint.data'
F_CHGT = '../scf/nfchgt.data'
... ...
入力パラメーターファイルはF_INPによってinput_band_Si.data を、k点のデータはF_KPOINTによってkpoint.dataであることを指定しています。
入力ファイル kpoint.data は、ツールband_kpoint.plを用いて生成します。band_kpoint.plのFCC用の入力ファイルは samples/tools/bandkpt_fcc_xglux.in です。
% ../../../../bin/band_kpoint.pl ../../../../tools/bandkpt_fcc_xglux.in
これらの入力ファイルを使って、プログラムekcal を実行します。
% mpirun ../../../../bin/ekcal
出力ファイル nfenergy.data から、ツール band.pl を用いて、 バンド構造図を作成します。
ツール band.plを以下のように実行すると、Postscript 形式のファイル band_structure.eps が作成されます。
% ../../../../bin/band.pl nfenergy.data ../../../../tools/bandkpt_fcc_xglux.in -erange=E1,E2 -with_fermi -color
この例題では、描画するエネルギー範囲の最小値 E1 と最大値 E2 を、以前同様 E1 = -13 と E2 = 5 として、 以下のように実行します。
% ../../../../bin/band.pl nfenergy.data ../../../../tools/bandkpt_fcc_xglux.in -erange=-13,5 -with_fermi -color
Si2のバンド構造を、図 5.6 に示します。

図 5.6 Si2のバンド構造
5.3. スピン分極を考慮した計算
強磁性体や反強磁性体を扱う場合にはスピン分極を考慮する必要があります。スピン分極の考慮した計算について説明します。
ここでは、強磁性の例として体心立方鉄を、反強磁性の例として体心立方クロムを利用して説明を行います。
5.3.1. 強磁性の計算
5.3.1.1. 入力パラメータ
強磁性の例として体心立方鉄を例に説明します。計算例題は、 samples/basic/bcc_Fe です。
Control{
condition = initial
cpumax = 3 hour
max_iteration = 250
}
accuracy{
cutoff_wf = 25 rydberg
cutoff_cd = 225.00 rydberg
num_bands = 20
ksampling{
method = mesh
mesh{ nx = 10, ny = 10, nz = 10 }
}
smearing{
method = tetrahedral
}
xctype = ggapbe
scf_convergence{
delta_total_energy = 1.e-10 hartree
succession = 3
}
}
structure{
unit_cell_type = Bravais
unit_cell{
#units angstrom
a = 2.845, b = 2.845, c = 2.845
alpha = 90, beta = 90, gamma = 90
}
symmetry{
crystal_structure = bcc
}
magnetic_state = ferro
atom_list{
atoms{
!#tag rx ry rz element
0.000 0.000 0.000 Fe
}
}
element_list{ !#tag element atomicnumber zeta dev
Fe 26 0.275 1.5 }
}
Postprocessing{
dos{
sw_dos = ON
method = tetrahedral
deltaE = 1.e-4 hartree
nwd_dos_window_width = 10
}
charge{
sw_charge_rspace = OFF
filetype = cube
title = "This is a title line for FM bcc Fe"
}
}
printlevel{
base = 1
}
結晶構造の指定
変数crystal_structureで、体心立方構造の結晶(bccという値)であることを指定しています。よって、ユニットセルはブラベー格子によって指定しているので原子は1つのみ記述しています。体心位置にある原子は指定していない点にご注意ください. crystal_structureにbccという値を指定すると、プログラムが指定の格子を基本格子に変換するので体心位置の原子の指定は不要となります。
スピン自由度の指定方法
強磁性体を扱う場合には, magnetic_state をferroと指定します。
structure{
magnetic_state = ferro !{para|antiferro|ferro}
}
さらに各原子のスピン分極の初期値を指定する必要があります。 入力ファイルにある
element_list{ #tag element atomicnumber zeta dev
Fe 26 0.275 1.5
}
の zeta = 0.275 という変数の値が, アップ・スピンとダウン・スピンの 密度の差を表す, スピン分極 \(\zeta = (n_{\uparrow} - n_{\downarrow})/(n_{\uparrow} + n_{\downarrow})\) の初期値を示しています.
5.3.1.2. 計算結果の出力
スピン分極の変化はログファイルoutput000に出力されます。以下のようにして確認することができます.
% grep charge output000 | grep NEW
!NEW total charge (UP, DOWN, SUM) = 5.02955985 (+) 2.97044015 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.03034164 (+) 2.96965836 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.04318734 (+) 2.95681266 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.05422913 (+) 2.94577087 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.07574696 (+) 2.92425304 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.10717707 (+) 2.89282293 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12471628 (+) 2.87528372 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12861238 (+) 2.87138762 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12847549 (+) 2.87152451 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12852226 (+) 2.87147774 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12859310 (+) 2.87140690 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12859664 (+) 2.87140336 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12859623 (+) 2.87140377 (=) 8.00000000
!NEW total charge (UP, DOWN, SUM) = 5.12859688 (+) 2.87140312 (=) 8.00000000
ここで, スピン分極の定義 \(\zeta = (n_{\uparrow} - n_{\downarrow})/(n_{\uparrow} + n_{\downarrow})\) を使うと, これが \(\zeta = 0.2821\) という値に収束していることが分かります.
5.3.2. 反強磁性の計算
反強磁性体の場合も、強磁性の計算と基本的には同じです。ただし、反強磁性を実現するためには初期スピン配置を反強磁性的にする必要があります。 そうしないと、高い確率で準安定状態である強磁性の解へ収束します。
強磁性の項においても説明したように、初期スピン分極は元素ごとにしか定義することができません。そこで、PHASEでは同じ擬ポテンシャルを利用する元素を複数用意し、各々にスピン分極を設定することによって反強磁性的な初期スピン配置を指定することができます。
5.3.2.1. 入力パラメータ
反強磁性の例として体心立方クロムを例として説明します。
ここでは、反強磁性秩序をスピン分極が異なる原子を異なる原子種として扱う(磁気秩序magnetic_stateはferroと指定する)方法を紹介します。Crの元素指定は、以下のようにCr1とCr2として指定します。
element_list{
#tag element atomicnumber zeta
Cr1 24 0.3
Cr2 24 -0.3
}
}
Cr1 とCr2 という2 種の元素を定義し、初期スピン分極としてそれぞれ0.3, -0.3という値を設定しています。原子座標は次のように設定します。これは初期値で、電子状態計算が進むに従いスピン分極の大きさはこの設定値から変化することに注意して下さい。
atom_list{
atoms{
#tag rx ry rz element
0.000 0.000 0.000 Cr1
0.500 0.500 0.500 Cr2
}
}
原点位置の原子Cr1に、体心位置の原子をCr2にしています
スピン自由度の指定として、magnetic_state をferroと指定します。
magnetic_state = ferro !{para|ferro} |
file_names.dataファイルでは、擬ポテンシャルを次のように指定します。
&fnames
F_INP = './nfinp.data'
F_POT(1) = '../../Cr_paw.pp'
F_POT(2) = '../../Cr_paw.pp'
/
Cr_paw.ppは、内容としてはCr元素の擬ポテンシャルファイルです。このような設定を施すことによって、Cr1, Cr2は別元素とみなされながら擬ポテンシャルは同じものが使用されることになります。
この方法を利用することによって、より複雑な磁気構造を持つ系の計算を行うことも可能です。
5.4. 構造最適化
原子に働く力を利用して、構造最適化を行うことができます。構造最適化機能の利用方法を説明します。
5.4.1. 入力パラメータ
構造最適化を行うには、入力ファイルを次のように記述します。
accuracy ブロックにおいて原子に働く力の最大値の指定を以下のように行います。このパラメータが、構造最適化の収束判定となります。
accuracy{
...
force_convergence{
max_force = 1.0e-3 hartree/bohr
}
...
}
max_forceのデフォルト値は、\(10^{- 3}\) hartree/bohrです。
structureブロックの原子の指定atom_listにmobile属性を定義し、最適化の対象となる原子に1 (もしくはyesないしon)という値を指定します。最適化の対象としない原子は0あるいは* (もしくはnoないしoff)とします。
...
structure{
...
atom_list{
!#tag element rx ry rz mobile
Ba 0.0000 0.5000 0.05 0
O 0.5000 0.0000 0.05 1
Ba 0.5000 0.0000 0.15 1
O 0.0000 0.5000 0.15 1
...
}
}
...
この例では、1番目のBa原子は最適化の対象とせず、2番目と4番目のO原子と3番目のBa原子が最適化の対象としています。
x, y, z座標を個別に最適化の対象とするかどうかを設定することも可能です。 この設定は、mobilex, mobiley, mobilez属性値によって行います。 mobilex, mobiley, mobilez属性値は、mobile属性値と同じ値がデフォルト値です。
...
structure{
...
atom_list{
!#tag element rx ry rz mobile mobilex
Ba 0.0000 0.5000 0.05 0 1
O 0.5000 0.0000 0.05 1 0
Ba 0.5000 0.0000 0.15 1 1
O 0.0000 0.5000 0.15 1 0
...
}
}
...
この例では、1番目のBa原子はx座標のみが、2番目および4番目のO原子はy座標とz座標のみが、3番目のBa原子はx, y, z座標が最適化の対象となります。
structure_evolution ブロックに、構造最適化の設定をします。
...
structure_evolution{
method = quench
dt = 50
...
}
...
method |
構造緩和の方法を指定します。 構造緩和のオプションとして、quench (quenched MD法) , cg2法(改良CG法;2がつかないcg方は非推奨) gdiis(GDIIS法), bfgs (BFGS法) , fire (FIRE法), lbfgs (LBFGS法)のいずれかが選べます。 デフォルト値はbfgsです。 |
dt |
構造緩和を行う際の時間刻みです。 quench法とfire法で用いられます。 大きい方が早く収束へ至りますが、大きすぎると計算を正しく進行させることができなくなる場合があります。 デフォルト値は原子単位で100です。 |
5.4.1.1. GDIIS, BFGS, LBFGS法の詳細設定
GDIIS法あるいはBFGS法は原子に働く力が大きい場合安定に計算できない場合があるので、力が大きい内はquenched MD法かCG法を利用し、ある程度力が小さくなってからGDIIS(BFGS)法に切り替える、という動作をします。GDIIS(BFGS)に切り替える前の最適化手法と切り替えの判定条件は、それぞれ変数initial_methodとc_forc2gdiisを利用して 次のように設定します.
...
structure_evolution{
method = gdiis
dt = 50
gdiis{
initial_method = cg2
c_forc2gdiis = 0.0025 hartree/bohr
}
}
...
ブロック名は、GDIIS, BFGS共通でgdiisです。デフォルト値はinitial_methodがcg2, c_forc2gdiisが0.05 hartree/bohr です。
5.4.1.2. FIRE法の詳細設定(バージョン2020.01以降)
methodにfireを指定するとFIRE法 (E. Bitzek F. G ähleret, M. Moseler, and P. Gumbsch, Physical Review Letters, 97 (2006) 170201) が使えます。FIRE法はquench法に似た手法ですが、時間刻みが可変となっている点に特徴があります。以下のように進行します。
原子間力Fと速度vとの内積Pを計算する。
vを \((1-\alpha) \cdot \bf{v} + \alpha \cdot \frac{\bf F}{|\bf F|} |{\bf V}|\) とする。
Pが0より大きく、かつPが負であったステップから \(N_{\rm min}\) ステップ以上経過しているのであれば時間刻みを大きくする。 \(\Delta t \cdot f_{\rm inc}\) と \(\Delta t_{\rm max}\) の内小さい方を採用する。 \(\alpha\) はファクター \(f_{\rm dec}\) をかけて小さくする。
Pが0以下の場合、タイムステップをファクター \(f_{\rm dec}\) をかけることによって小さくする。また、速度を0とし、 \(\alpha\) を既定値 \(\alpha_{\rm start}\) に設定する。
定性的に説明すると、「正しい方向(P > 0)に進んでいる限り時間刻みを増やし、勾配よりも速度を優先する」アルゴリズムです。FIRE法のパラメーターは、\(\alpha_{\rm start}, N_{\rm min}, f_{\rm inc}, \Delta t_{\rm max}, f_{\alpha}, f_{\rm dec}\) です。入力パラメーターファイルにおいて、以下の要領で設定できます。
structure_evolution{
fire{
incre_factor = 1.2
decre_factor = 1/1.2
decre_factor_alpha = 1/1.2
alpha_start = 1
nmin = 3
dtmax = 300
initial_dt = 100
invmass_factor = 2.e-5
}
}
incre_factorが \(f_{\rm inc}\) , decre_factorが \(f_{\rm dec}\) , decre_factor_alphaが \(f_{\alpha}\) , nminが \(N_{\rm min}\) , dtmaxが \(\Delta t_{\rm max}\) , alpha_startが \(\alpha_{\rm start}\) に対応します。また、initial_dtは初期の時間刻みです。invmass_factorには質量の逆数に相当する数値を指定します。デフォルト値は、上述の例で指定している値です。
5.4.1.3. limited-memory BFGS法による構造最適化 (バージョン2021.01以降)
概要
PHASE/0には構造最適化手法としてquench法、CG法、GDIIS法、BFGS法、FIRE法などが搭載されています。バージョン2021.01以降、文献 [Hjorth17] のlimited-memory BFGS法 (l-BFGS法)が選択肢に加わりました。 通常のl-BFGS法のほか、文献において提案されている前処理を施すこともできるようになっています。
l-BFGS法について
limited-memory BFGS法とは、BFGS法を効率化した最適化手法です。ヘッセ行列をメモリーに明示的に持つのではなく、iterationごとに構築していくことに特徴を持つ手法です。ヘッセ行列をあらわに持たないため、メモリー要求が小さいことが名称のlimited memoryの由来です。そのアルゴリズムの擬コードは、以下のように記述することができます。
ここで \(x_k, g_k`は :math:`k\) 回目のiterationにおける座標データとエネルギーの座標微分、 \(H_k^0\) は初期ヘッセ行列の推定値に相当する行列です。 その行列要素は (5.1) のように \(H_k^0 = \gamma_k I\) と計算することが一般的ですが、これを以下のような前処理行列で置き換えることが文献では提案されています。
ここで \(A, \mu, r_{nn}, r_{\rm cut}\) は任意に設定できるパラメーターです。 文献によると \(A\) はあまり結果を左右しないようです。文献でも採用されている3という値をデフォルト値としています。 \(r_{nn}\) は最近接間距離の最大値であり、 原子配置から自動的に決まる値です。 \(r_{\rm cut}\) はカットオフ距離に相当します。典型的には \(r_{nn}\) の2倍という値にります。 最も結果を左右するパラメーターが \(\mu\) です。このパラメーターは小さい方が速い収束が見込めますが、小さくし過ぎるとロバスト性が損なわれてしまいます。 PHASE/0のデフォルトの振る舞いとしては、前処理行列の行列要素の大きさが結果としておおむね通常のl-BFGS法の \(\gamma_k\) と同程度の値になるような値が採用されます。
使い方
構造最適化の設定は、入力パラメーターファイルのstructure_evolutionブロックにおいて行います。l-BFGS法を用いる場合、典型的には下記のような指定になります。
structure_evolution{
method = lbfgs
lbfgs{
c_iteration2gdiis=3
c_forc2gdiis = 0.05
gdiis_box_size = 6
initial_method = cg2
maxstep = 0.1
}
}
タグmethodにlbfgsを指定するとl-BFGS法を利用することができます。Limited-memory BFGS法(および通常のBFGS法、GDIIS法)の詳細設定は、structure_evolutionブロックの下のlbfgsブロック(bfgs, gdiisも可)において行うことができます。lbfgsブロック(もしくはbfgs, gdiisブロック)において設定できる主なパラメーターは下記の通り。
タグ名 |
説明 |
c_iteration2gdiis |
lbfgs法、bfgs法、gdiis法は計算の最初期から適用されるわけではなく、はじめは閾値を満たすまでは別の最適化手法が用いられます。閾値を満たしたあと、このタグの指定の回数経てからlbfgs法 (もしくはbfgs法、gdiis法)にに移行します。デフォルト値は3. |
c_forc2gdiis |
lbfgs法、bfgs法、gdiis法へ移行する際の閾値を力の単位で指定します。デフォルト値は0.05 hartree/bohr |
gdiis_box_size |
lbfgs法、bfgs法、gdiis法の履歴の大きさ。デフォルト値は6. |
initial_method |
lbfgs法、bfgs法、gdiis法へ移行する前の構造最適化手法を指定します。デフォルト値はcg2. |
maxstep |
lbfgs法において、最適化1回で原子が動ける距離の最大値を長さの単位で指定します。デフォルト値は0.1 bohr. この設定値はlbfgs法の場合のみ利用できます。 |
多くのタグ名がGDIIS法にちなんだものとなっているのは、本機能がGDIIS法の履歴活用手法を用いているためです。
Limited-memory BFGS法は、上述の通り前処理行列を作用させることができます。その設定は、下記の要領で行います。
structure_evolution{
method = lbfgs
...
sw_prec = on
prec{
A = 3.0
mu = -1
}
}
structure_evolutionブロックにおいてsw_prec = onとすると前処理が有効になります。前処理の詳細設定はprecブロックにおいて行います。設定できるパラメーターは下記の通り。
タグ名 |
説明 |
A |
|
mu |
|
例題
PHASE/0の構造最適化の例題は、 samples/structural_evolution 以下に用意されています。
すべての例題についてquench, bfgs, cg2, gdiisの例題が用意されていますが、ここにlbfgsディレクトリーを追加しました。
PHASE/0付属の例題を用いて、CG2, BFGS, l-BFGS法が収束するまでに要したionic iterationの回数を以下に報告します。
系 |
CG2 |
BFGS |
l-BFGS |
Si(001)面 |
84 |
102 |
63 |
SiO 2 結晶 |
15 |
15 |
11 |
TiO 2 結晶 |
21 |
31 |
17 |
ジクロロシクロヘキサン |
77 |
60 |
45 |
この表から、おおむねl-BFGS法が高速であることが分かります。必ずよい結果が得られるとは限りませんが、多くの問題でここで見たような傾向が見られており、 l-BFGS法は有力なオプションであると言えます。
Ask Hjorth Larsen et al. J. Phys.: Condens. Matter 29 (2017) 273002.
5.4.1.4. mobile属性値を“特定の原子位置もしくは任意の位置からの距離以内”の原子という形式で指定する方法(バージョン2020.01以降)
バージョン2020.01以降、“ある位置からある距離以内の原子をmobileな原子とする”設定が可能となりました。このような指定方法は、たとえば欠陥からxx Å以内の原子をmobileにしたい、という場合に便利です。このスタイルの設定は、以下の要領で行います。
structure{
sw_mobility_by_distance = on
mobility_by_distance{
target_atom = 66
target_posx = 0.2
target_posy = 0.2
target_posz = 2
distance = 5 angstrom
}
}
structureブロックの下のsw_mobility_by_distanceをonとするとこの設定方法が利用できます。この場合はatomsテーブルにおける指定は無効になる点には注意してください。この設定方法の詳細はmobility_by_distanceブロックにおいて設定します。target_atomに中心にしたい原子のIDを指定します。この数値が設定されている場合原子位置が、されていない場合はユーザー指定の位置が中心となります。target_posx, target_posy, target_poszによって原子中心でない場合の位置のx, y, z座標を指定します。デフォルト値はいずれも0です。distanceで、中心からの距離を指定します。この例の場合中心から5 Å以内に存在する原子はmobile = on, その外の原子はmobile = offと設定されます。
5.4.1.5. mobile属性値についての注意
mobile属性は、その名称から原子座標を動かすか・動かさないかを指定するもののように思えますが、これは場合と考え方(座標系)によります。 たとえば、格子を含む最適化を行う場合、mobile = offの原子も格子の変形にあわせてカルテシアン座標は変化します。 この場合不変となるのは相対座標です。
5.4.2. 計算結果の出力
構造最適化を施すと、F_ENFファイル(既定のファイル名:nfefn.data)にエネルギーや原子に働く力の最大値の履歴が、F_DYNMファイル(既定のファイル名:nfdynm.data)に原子配置の履歴が出力されます。
5.4.3. 計算例:シリコン結晶の構造最適化
シリコン結晶の構造最適化の計算例です。安定な原子配置から原子位置をずらして、そこからの緩和過程を計算する例題です。
計算例題は、 samples/basic/Si2/relax です。
入力パラメーターファイル
ファイル file_names.data の中では、入力パラメーターファイル input_relax_Si.dataと、原子の位置座標と各原子に働く力の 計算結果の出力ファイル nfdynm.data が指定されています。
F_INP = './input_relax_Si.data'
...
F_DYNM = './nfdynm.data'
...
入力パラメーターファイルinput_relax_Si.dataは、格子間隔を0.125ではなく0.130 とし、安定な原子配置から原子位置をずらしています。 また、mobile 変数の値を yes にして、原子位置を可変にしています。
structure{
...
atom_list{
atoms{
#tag rx ry rz element mobile
0.130 0.130 0.130 Si yes
-0.130 -0.130 -0.130 Si yes
}
}
}
accuracyブロックで原子に働く力の収束条件を指定します。
accuracy{
force_convergence{
max_force = 1.0e-3
}
}
計算結果
計算結果の出力ファイル nfdynm.data は以下の通りです。
#
# a_vector = 0.0000000000 5.1300000000 5.1300000000
# b_vector = 5.1300000000 0.0000000000 5.1300000000
# c_vector = 5.1300000000 5.1300000000 0.0000000000
# ntyp = 1 natm = 2
# (natm->type) 1 1
# (speciesname) 1 : Si
#
cps and forc at (iter_ion, iter_total = 1 14 )
1 1.333800000 1.333800000 1.333800000 -0.011489 -0.011489 -0.011489
2 -1.333800000 -1.333800000 -1.333800000 0.011489 0.011489 0.011489
cps and forc at (iter_ion, iter_total = 2 21 )
1 1.322311395 1.322311395 1.322311395 -0.009145 -0.009145 -0.009145
2 -1.322311395 -1.322311395 -1.322311395 0.009145 0.009145 0.009145
cps and forc at (iter_ion, iter_total = 3 30 )
1 1.277473011 1.277473011 1.277473011 0.001802 0.001802 0.001802
2 -1.277473011 -1.277473011 -1.277473011 -0.001802 -0.001802 -0.001802
cps and forc at (iter_ion, iter_total = 4 36 )
1 1.279275408 1.279275408 1.279275408 0.001305 0.001305 0.001305
2 -1.279275408 -1.279275408 -1.279275408 -0.001305 -0.001305 -0.001305
cps and forc at (iter_ion, iter_total = 5 43 )
1 1.284010642 1.284010642 1.284010642 0.000017 0.000017 0.000017
2 -1.284010642 -1.284010642 -1.284010642 -0.000017 -0.000017 -0.000017
このうち、# 記号で始まる部分は入力データの一部を表していますが、その次の行は、 イオンすなわちコア原子の位置座標を一回更新する間に、全更新回数が14回であったこと、 すなわち、この間に波動関数が13回更新されたことを示しています。 波動関数の更新に対する収束条件は、これまでの例題と同様に、 全エネルギーに対して課されています。
また、その次の2行は、原子の番号、原子位置(x,y,z, bohr単位)、 および力の成分 (x,y,z, hartree/bohr単位)の計算結果を表しています。 これにより、結果を下まで辿っていくと、計算が進むにつれて、原子に働く力が急激に 減少していくことが分かります。 最後の更新で、力の各成分の計算結果が、最初に指定された収束条件以下になったために、 緩和過程の計算が終了しています。
5.5. 表面の計算
5.5.1. 表面の計算を実行するには
PHASEは系に周期境界条件を課す必要があるので、厳密な意味では表面などの有限系を扱うことはできません。しかし、充分な“真空層”を設けることにより、事実上表面と変わらない系を扱うことは可能です。真空層は、底面と表面が相互作用しない程度の大きさを取ります。通常、10Å以上の真空層を採用します。
水素終端されたシリコン表面の計算を例とします。 入力ファイルは samples/surface/H_Si001_p2x1 以下にあります。この構造の計算には、 図 5.7
に示されるようなスラブ模型を使います。スラブの下側のSi原子のボンドは、仮想的な水素原子で終端しています。
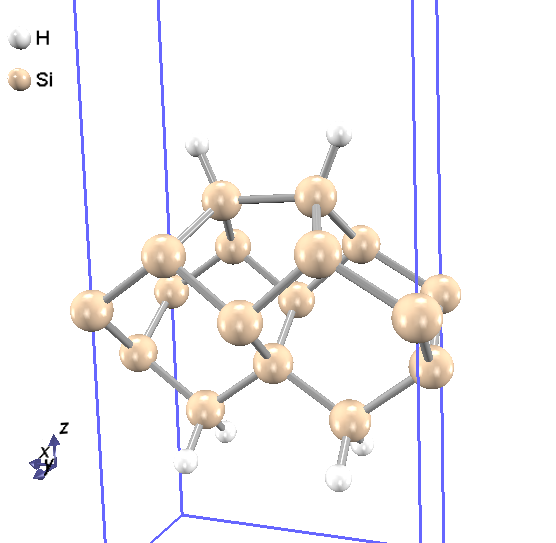
図 5.7 水素終端したSi(001)-p(2×1) 表面の構造図
この例で使用するfile names.data です。
&fnames
F_INP = './input_SiH2x1.data'
F_POT(1) = '../../pp/Si_ggapbe_paw_nc_01m.pp'
F_POT(2) = '../../pp/H_ggapbe_paw_nc_01m.pp'
................................
/
F_POT(1) と F_POT(2) に、Si 原子と H原子の擬ポテンシャルを指定しています。
入力パラメータ例です。
以下はカットオフエネルギーやk点サンプリングの指定です。
accuracy{
cutoff_wf = 20.00 rydberg
cutoff_cd = 80.00 rydberg
num_bands = 28
ksampling{
method = monk ! {mesh|file|directin|gamma}
mesh{ nx = 2, ny = 4, nz = 1 }
kshift{ k1 = 0.5, k2 = 0.5, k3 = 0.0 }
}
...........................
}
この例では、スラブ模型を用いているため、k点のサンプリングは\(k_{z}\) 方向には1点だけを取っています。
以下は座標データの指定です。
structure{
unit_cell_type = primitive
unit_cell{
a_vector = 14.512 0.000 0.000
b_vector = 0.000 7.256 0.000
c_vector = 0.000 0.000 30.784
}
symmetry{}
magnetic_state = para !{para|af|ferro}
atom_list{
coordinate_system = internal
atoms{
#default weight = 1, element = Si, mobile = 0
#tag rx ry rz element
0.26177 0.50000 0.65651 H
0.73823 0.50000 0.65643 H
0.34138 0.50000 0.56971
0.65858 0.50000 0.56966
0.26229 0.00000 0.49388
0.73763 0.00000 0.49385
0.00000 0.00000 0.41498
0.50000 0.00000 0.40298
0.00000 0.50000 0.32769
0.50000 0.50000 0.32150
0.25000 0.50000 0.24167
0.75000 0.50000 0.24167
0.25000 0.20000 0.18269 H
0.25000 0.80000 0.18269 H
0.75000 0.20000 0.18269 H
0.75000 0.80000 0.18269 H
}
}
}
postprocessing{
charge{
sw_charge_rspace = ON
filetype = cube !{cube|density_only}
title = "Si(001) p(2x1) surface terminated by H atoms"
}
}
atoms の中で、デフォルト値として元素名を Si に設定しているので、 変数 element に H と入力している以外の原子の元素名は Si になります。 また、やはりデフォルト値として mobile = 0 としているので、全ての原子の座標位置は固定されています。
grepコマンドを用いて全エネルギーの収束状況を確認すると、以下のような結果が得られます。
% grep TH output000
TOTAL ENERGY FOR 1 -TH ITER= -40.800374098495 EDEL = -0.408004D+02 : SOLVER = MATDIAGON : Charge-Mixing = PULAY
TOTAL ENERGY FOR 2 -TH ITER= -42.619401425789 EDEL = -0.181903D+01 : SOLVER = SUBMAT + PDAVIDSON : Charge-Mixing = PULAY
TOTAL ENERGY FOR 3 -TH ITER= -42.771292215127 EDEL = -0.151891D+00 : SOLVER = SUBMAT + PDAVIDSON : Charge-Mixing = PULAY
TOTAL ENERGY FOR 4 -TH ITER= -42.778649035692 EDEL = -0.735682D-02 : SOLVER = SUBMAT + PDAVIDSON : Charge-Mixing = PULAY
TOTAL ENERGY FOR 5 -TH ITER= -42.781674463294 EDEL = -0.302543D-02 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 6 -TH ITER= -42.787305265390 EDEL = -0.563080D-02 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 7 -TH ITER= -42.790915993227 EDEL = -0.361073D-02 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 8 -TH ITER= -42.792248833909 EDEL = -0.133284D-02 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 9 -TH ITER= -42.791249566593 EDEL = 0.999267D-03 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 10 -TH ITER= -42.791828775520 EDEL = -0.579209D-03 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 11 -TH ITER= -42.792299736444 EDEL = -0.470961D-03 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 12 -TH ITER= -42.792825149910 EDEL = -0.525413D-03 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 13 -TH ITER= -42.792904492271 EDEL = -0.793424D-04 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 14 -TH ITER= -42.792903178943 EDEL = 0.131333D-05 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 15 -TH ITER= -42.792904585521 EDEL = -0.140658D-05 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 16 -TH ITER= -42.792920290125 EDEL = -0.157046D-04 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 17 -TH ITER= -42.792926361303 EDEL = -0.607118D-05 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 18 -TH ITER= -42.792927175935 EDEL = -0.814632D-06 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 19 -TH ITER= -42.792927686841 EDEL = -0.510906D-06 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 20 -TH ITER= -42.792927748178 EDEL = -0.613377D-07 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 21 -TH ITER= -42.792928070956 EDEL = -0.322778D-06 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 22 -TH ITER= -42.792928324357 EDEL = -0.253400D-06 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 23 -TH ITER= -42.792928345097 EDEL = -0.207401D-07 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
TOTAL ENERGY FOR 24 -TH ITER= -42.792928350400 EDEL = -0.530343D-08 : SOLVER = SUBMAT + RMM3 : Charge-Mixing = PULAY
この例題は、固体表面構造に対するエネルギー計算だけを目的にしていますが、もし原子位置の緩和過程の計算を行う場合は、以下のように、下端の仮想水素とそれらと結合した Si 原子を固定し、それら以外の原子を可動 (mobile = 1) に変えてやる必要があります。
atoms{
#default weight = 1, element = Si, mobile = 1
#tag rx ry rz element mobile
0.26177 0.50000 0.65651 H
0.73823 0.50000 0.65643 H
0.34138 0.50000 0.56971
0.65858 0.50000 0.56966
0.26229 0.00000 0.49388
0.73763 0.00000 0.49385
0.00000 0.00000 0.41498
0.50000 0.00000 0.40298
0.00000 0.50000 0.32769
0.50000 0.50000 0.32150
0.25000 0.50000 0.24167 * 0
0.75000 0.50000 0.24167 * 0
0.25000 0.20000 0.18269 H 0
0.25000 0.80000 0.18269 H 0
0.75000 0.20000 0.18269 H 0
0.75000 0.80000 0.18269 H 0
}
Si(001) 表面のバックルしたダイマーの安定構造は p\((2 \times 1)\) ではなく c\((4 \times 2)\) ですが、この構造を再現するには、Si ダイマーをもう一つ増やすなどして、 最上層に位置する Si ダイマーの総数を偶数個にしなければなりません。
5.5.2. 反転対称性を考慮した表面の計算
表面には、反転対称性がある場合があります。反転対称性を利用することによって、ほぼ同等の計算負荷で2
倍の厚さの表面モデルを取り扱うことが可能です。Pt 表面の(111)
面を例とします。入力ファイルは samples/surface/Pt 以下のサブディレクトリーに配置されています。
この例題の入力ファイルのstructureブロックは以下のようになっています。
structure{
element_list{
#tag element atomicnumber mass
Pt 78 355606.909
}
atom_list{
atoms{
#units angstrom
#tag element rx ry rz mobile weight
Pt 0.0 0.0 0.0 * *
Pt 0.0 0.5 0.0 * *
Pt 0.5 0.0 0.0 * *
Pt 0.5 0.5 0.0 * *
Pt 0.6666666666666667 0.6666666666666666 0.05370700299352444 * 2
Pt 0.6666666666666667 0.16666666666666674 0.05370700299352444 * 2
Pt 0.16666666666666685 0.6666666666666666 0.05370700299352444 * 2
Pt 0.16666666666666669 0.16666666666666674 0.05370700299352444 * 2
Pt 0.3333333333333333 0.33333333333333337 0.10741400477864135 * 2
Pt 0.3333333333333333 0.8333333333333334 0.10741400477864135 * 2
Pt 0.8333333333333333 0.33333333333333337 0.10741400477864135 * 2
Pt 0.8333333333333333 0.8333333333333334 0.10741400477864135 * 2
Pt 0.0 0.0 0.16112100656375825 * 2
Pt 0.0 0.50 0.16112100656375825 * 2
Pt 0.50 0.0 0.16112100656375825 * 2
Pt 0.50 0.50 0.16112100656375825 * 2
Pt 0.666666666666667 0.6666666666666667 0.21482800834887514 * 2
Pt 0.666666666666667 0.1666666666666667 0.21482800834887514 * 2
Pt 0.16666666666666663 0.6666666666666667 0.21482800834887514 * 2
Pt 0.16666666666666669 0.1666666666666667 0.21482800834887514 * 2
Pt 0.3333333333333336 0.3333333333333335 0.26853501013399206 on 2
Pt 0.33333333333333354 0.8333333333333335 0.26853501013399206 on 2
Pt 0.8333333333333335 0.3333333333333335 0.26853501013399206 on 2
Pt 0.8333333333333333 0.8333333333333335 0.26853501013399206 on 2
Pt 0.0 0.0 0.3222420119191089 on 2
Pt 0.0 0.50 0.3222420119191089 on 2
Pt 0.50 0.0 0.3222420119191089 on 2
Pt 0.50 0.50 0.3222420119191089 on 2
Pt 0.666666666666667 0.666666666666667 0.37594901370422584 on 2
Pt 0.666666666666667 0.166666666666667 0.37594901370422584 on 2
Pt 0.166666666666667 0.666666666666667 0.37594901370422584 on 2
Pt 0.166666666666667 0.166666666666667 0.37594901370422584 on 2
}
}
unit_cell{
#units angstrom
a_vector = 5.6568542495 0.00 0.00
b_vector = 2.8284271247 4.8989794856 0.00
c_vector = 0.00 0.00 43.00
}
symmetry{
tspace{
lattice_system = primitive
}
method = automatic
sw_inversion = on
}
unit_cell_type = bravais
}
weight属性として2という値が振られた原子がありますが、これは原点を中心とした反転対称位置にも原子を配置するという指定に対応します。 この例題では、原点を中心に反転対称性があるため、それを活用するために、symmetryブロックの下のsw_inversion変数をonとしています。 この座標データを可視化すると、図 5.8 となります。
この例のように、表面は厚さ方向の中央を原点とすることによって反転対称性があるようになる場合があります。このような場合は、sw_inversionパラメータをonとすることによって計算量を削減することができます。表面にさらに分子や原子などを吸着させた計算を行う場合は、両側の反転対称位置に配置することによってやはり反転対称性を持たせることが可能です。
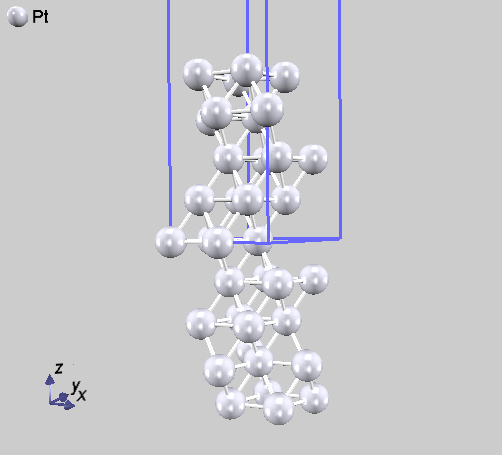
図 5.8 Pt(111)面の原子配置。表面モデルの中央を原点にすることによって反転対称性がある。
5.5.3. 計算例:金属表面の生成エネルギー
0Kにおける表面の生成エネルギーは、以下のように評価することが可能です。
ここで\(\gamma\)が表面生成エネルギー、\(E_{s}\)が表面の全エネルギー、\(E_{b}\)が対応する結晶の全エネルギー、\(A\)が表面積です。2Aで割っているのは、計算では表面が2つ現れるからです。 また、\(E_{b}\)は表面モデルと原子数が合うようにスケールしたあとで差を評価します。
反転対称性を考慮した表面の計算例は、白金表面の生成エネルギーの計算です。
Pt(111) 面 |
9層の(111)面, 計36原子。 格子定数は、\(a = b = 5.657Å,c = 30Å ,\alpha = \beta = 90^{\circ},\gamma = 120^{\circ}\) 図 5.8 のモデル |
Pt(110)面MR |
15層のmissing-row (MR) (110)面, 計28原子 MR面とは、表面の“列”をなしている原子が1列おきに欠けている表面のモデル。 格子定数は\(a=4Å,\) \(b = 2.828427125Å,\) \(c = 30Å,\) \(\alpha = \beta = \gamma = 90^{\circ}\) 図 5.10 のモデル(この図では、スーパーセルで表示している)。 |
Pt(110)面 |
15層の(110)面, 計15原子。 格子定数は \(a = 8Å,b = 2.8284271248Å\) \(,c = 30Å\), \(\alpha = \beta = \gamma = 90^{\circ}\) 図 5.9 のモデル(この図では、スーパーセルで表示している)。 |
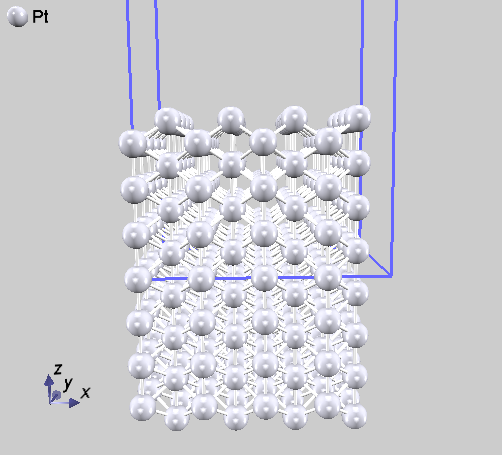
図 5.9 Pt(110) 理想表面(スーパーセル表示)
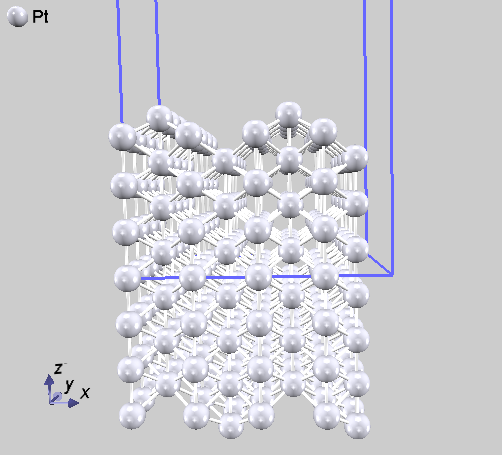
図 5.10 Pt(110) 面missing-row 構造(スーパーセル表示)
白金表面は、(111)面が最も安定で、(110)面についてはmissing-row (MR) 再配列が成されるとされています。このようなことが、表面生成エネルギーの計算から再現できることを確認します。
主な計算条件です。
いずれのモデルも反転対称性を考慮
カットオフエネルギーは25 Rydberg
k 点サンプリングは、(111) に対しては6x6x1, (110) に対しては6x8x1, (110) MR に対しては3x8x1
構造最適化はBFGS法によって実施;力の収束判定は\(2 \times 10^{- 4}\) hartree/bohr
構造最適化の対象となる原子は、最表面から4 層ずつ
このようにして得られた表面生成エネルギーの計算結果を、 表 5.6 にまとめました。(111)面の生成エネルギーが小さく、次に(110) MR, 最も生成エネルギーが大きいのが(110)面という結果が得られました。
|
|||
|---|---|---|---|
生成エネルギー (eV/Å2) |
0.089 |
0.099 |
0.108 |
5.6. 原子・分子の計算
原子・分子の計算は、真空層を設けることによって行います。原子や分子の場合は、周期的境界条件の影響がないように、すべてのセルベクトルの方向で真空層を設ける必要があります。 通常、\(k\)サンプリングは\(\Gamma\)点のみを利用します。
5.6.1. 入力パラメータ
原子・分子の計算は、真空層を設けるようにunit_cellを指定します。
unit_cell{
a_vector = 15.0 0.0 0.0
b_vector = 0.0 15.0 0.0
c_vector = 0.0 0.0 15.0
}
水分子の計算の入力パラメータです。原子座標に対し、十分に大きなユニットセルとしています。
Control{
condition = initial
cpumax = 1 day ! maximum cpu time
max_iteration = 6000
}
accuracy{
cutoff_wf = 25.00 rydberg
cutoff_cd = 225.00 rydberg
num_bands = 8
xctype = ggapbe
initial_wavefunctions = matrix_diagon
matrix_diagon {
cutoff_wf = 5.0 rydberg
}
ksampling{
method = gamma
}
scf_convergence{
delta_total_energy = 1.e-10
succession = 3
num_max_iteration = 300
}
force_convergence{
max_force = 1.e-4
}
initial_charge_density = Gauss
}
structure{
unit_cell_type = primitive
unit_cell{
a_vector = 15.0 0.0 0.0
b_vector = 0.0 15.0 0.0
c_vector = 0.0 0.0 15.0
}
symmetry{
tspace{
lattice_system = primitive
generators{
#tag rotation tx ty tz
C2z 0 0 0
IC2x 0 0 0
}
}
}
atom_list{
coordinate_system = cartesian
atoms{
!#default mobile=on
!#tag rx ry rz element
-1.45 0.000 1.123 H
1.45 0.000 1.123 H
0.0 0.0 0.0 O
}
}
element_list{ #units atomic_mass
#tag element atomicnumber zeta dev
H 1 1.00 0.5
O 8 0.17 1.0 }
}
wavefunction_solver{
solvers {
!#tag sol till_n dts dte itr var prec cmix submat
msd 5 0.1 0.1 1 tanh on 1 on
lm+msd 10 0.1 0.4 50 tanh on 1 on
rmm3 -1 0.4 0.4 1 tanh on 2 on
}
rmm {
edelta_change_to_rmm = 1.d-6
}
lineminimization {
dt_lower_critical = 0.1
dt_upper_critical = 3.0
}
}
charge_mixing{
mixing_methods {
!#tag id method rmxs rmxe itr var prec istr nbxmix update
1 broyden2 0.3 0.3 1 linear on 5 10 RENEW
2 simple 0.2 0.5 100 linear on * * *
}
}
5.7. 電荷密度の出力
PHASEはSCF計算中は逆空間で電荷密度を扱いますが、収束した電荷密度を実空間に逆フーリエ変換し、出力させることも可能です。こうすることによってPHASE-Viewerなどを利用して電荷密度の可視化を行うことが可能です。電荷密度を実空間に出力させるためには、入力ファイルの最上位にpostprocessingブロックを作成し、さらにその下にchargeブロックを作成しその下で設定を行います。
postprocessing{
charge{
sw_charge_rspace = on
filetype = cube
}
}
chargeブロックの下では以下の変数の設定を行います。
sw_charge_rspace |
電荷密度を実空間で出力するかどうかを指定する真偽値です。 onにすると実空間の電荷密度が出力されます。 |
filetype |
電荷密度データのデータフォーマットを指定します。 density_onlyとcubeが選べます。 density_onlyの場合電荷密度のみが出力されます。 デフォルト値はdensity_onlyです。 cubeの場合、Gaussian Cube形式で電荷密度が出力されます。このパラメーターは、cubeに設定することを推奨します。 |
title |
Gaussian Cubeファイルの“見出し”を指定します。 空白文字を含める場合、全体を半角の2重引用符で囲みます。 |
また、filetypeとしてcubeを選択した場合、file_names.dataファイルにおいて電荷密度ファイルのファイル名を変更しておくことを推奨します。
&fnames
...
F_CHR = './nfchr.cube'
/
変更しない場合のデフォルト値はnfchr.dataです。
スピン分極を考慮している場合は、file_names.dataで指定したファイル名がnfchr.cubeであったとすると、nfchr.up.cubeとnfchr.down.cubeという2つのファイルにそれぞれスピンアップ・ダウン に対応する電荷密度データが出力されます。参考のため、 図 5.11 に鉄の多数派スピンと少数派スピンの電荷密度をPHASE-Viewerで可視化した様子を示します。
さらに、特定のエネルギー範囲の電荷密度を抜き出して出力させる機能もPHASEには備わっています。この機能については、応用機能において解説します。
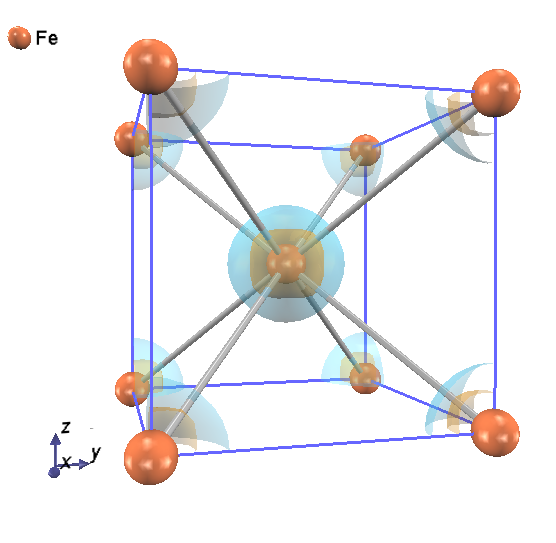
図 5.11 Fe の電荷密度分布図. 青色とオレンジ色の面は, 自発磁化により生じた, 多数派スピンと小数派スピンによる 電荷密度分布の等値面を表す.
5.8. 波動関数の出力
電荷密度と同じように波動関数を実空間にマップしたボリュームデータとして出力することができます。得られた結果はPHASE-Viewerなどを利用して可視化することができます。波動関数を出力させるためにはPostprocessingブロックを作成し、さらにその下にwfブロックを作成しその下で設定を行います。
postprocessing{
wf{
sw_wf_rspace = on
filetype = cube
eigmin = 0.13 hartree
eigmax = 0.14 hartree
}
}
sw_wf_rspace = onとすることによってこの機能が有効になります。filetype = cubeとするとcube形式でファイルが出力されます。eigmin, eigmaxに出力する波動関数の固有値の範囲を指定します。これはフェルミエネルギーからみた相対値ではなく絶対値を用います。デフォルト値はeigmin = -100 Ha, eigmax = 100 Haで、事実上すべての準位が対象となります。
file_names.dataファイルには波動関数ファイルのファイル名を指定します。デフォルト値はnfwfk.dataですが、cube形式で出力する場合拡張子をcubeに変更することが推奨されます。以下のように記述します。
&fnames
F_WFk = 'nfwfk.cube'
/
実際に得られるcubeファイルは上述のファイル名の接頭部分に準位にちなんだ文字列が付与されたものです。具体的には、nfwfk.kxxxxnyyyyy.cube というファイル名になります。xxxx がk点の指標、yyyyy がバンドの指標です。得られるcubeファイルは分子軌道形式のcubeファイルとなっており、1つのファイルに波動関数の実部と虚部両方が記録されます。
Cube形式で出力した場合wfsq.pyスクリプトを利用することによって波動関数の二乗を出力させることができます。以下のように利用することができます。
$ $HOME/phase0_2025/bin/wfsq.py --input="nfwfk*.cube" --output=nfwfsq
次のオプションを用いることができます。
-i INPUT, --input=INPUT |
入力のCubeファイルを指定します。指定にはワイルドカードを使うことができます。ただし、*や?などの特殊な文字を用いる場合引用符または2重引用符で文字列を囲います。また、カンマ区切りによって複数の文字列を指定することもできます。デフォルト値は*.cubeです |
-o PREFIX, --output_prefix =PREFIX |
出力ファイルの接頭辞を指定します。デフォルト値はnfwfsq |
-a, --append |
結果を入力のcubeファイルに追記したい場合に指定するオプションです。この場合PREFIXは意味を成しません |
5.9. 状態密度の計算
SCF計算が収束したのち、状態密度の計算を行わせることができます。電荷密度の計算を行うためには、入力ファイルの最上位にpostprocessingブロックを作成し、さらにその下にdosブロックを作成しその下で設定を行います。
postprocessing{
dos{
sw_dos = on
method = gaussian
deltaE_dos = 1e-4 hartree
}
}
dos ブロックでは以下の設定を行うことができます。
sw_dos |
状態密度計算を行うかどうかを指定する真偽値です。 状態密度の計算を行う場合onとします。 |
method |
状態密度の計算方法を指定します。 gaussianとtetrahedralのいずれかを選択することができます。gaussianを選択した場合、エネルギー準位をガウス関数によって幅を持たせた上で計算した状態密度が得られます。tetrahedralの場合四面体法による高精度な状態密度計算を行うことができます。ただしtetrahedralを利用する場合後述の四面体法が利用できる条件もご参照ください。 |
deltaE_dos |
状態密度計算に利用されるエネルギーの幅をハートリー単位で指定します。デフォルト値は1e-4 hartreeです。 |
状態密度の計算方法としてtetrahedralを利用する場合、以下の条件が満たされている必要があります。
k点サンプリング手法としてmesh 法を採用している
accuracy{
ksampling{
method = mesh
}
}
smearing の方法としてtetrahedral 法を採用している
accuracy{
smearing{
method = tetrahedral
}
}
以上が満たされていないとgaussian 法による状態密度計算が行われてしまうので、ご注意ください。
参考のため、gaussian 法とtetraheral 法で計算した体心立方鉄の状態密度をそれぞれ 図 5.12 と 図 5.13 に示します。k 点メッシュはそれぞれ10 × 10 × 10 を採用しました。 Tetrahedral法で計算状態密度の方がシャープで精度のよいものが得られていることが分かります。

図 5.12 Gaussian 法で計算した体心立方鉄の状態密度

図 5.13 Tetrahedral 法で計算した体心立方鉄の状態密度
PHASEには原子や層によって分割した“局所状態密度”を計算する機能も備わっています。この機能については6 章 において説明します。
5.10. バンド構造の計算
5.10.1. k 点のデータの作成
バンド構造の計算には、バンド分散を計算するk点のデータが必要です。
k点のデータは、ツールband_kpoint.plを利用して作成します。まずband_kpoint.pl用の入力ファイルを作成します。その形式は、以下のようなものです。
dkv
b1x b2x b3x
b1y b2y b3y
b1z b2z b3z
n1 n2 n3 nd # Symbol
...
dkvが\(k\)点の間隔, b1x,b1y,b1zは逆格子ベクトル\(b_{1}\)のx,y,z成分です。 逆格子ベクトル\(b_{2}\),\(b_{3}\)についても同様です。 五行目以降に特殊\(k\)点とそのシンボルの指定をします。 シンボルの指定は必須ではありませんが、指定がある場合バンド構造図作成の際に利用されます。 整数\(n_{1},n_{2},n_{3},n_{d}\)を用いて\(k\)ベクトルを
のように指定します。シンボルは#の後に書いてください。 面心立方格子の場合の例を示します。
0.02 <---- k点の間隔
-1.0 1.0 1.0
1.0 -1.0 1.0
1.0 1.0 -1.0
0 1 1 2 # X <---- n1 n2 n3 nd # Symbol
0 0 0 1 # {/Symbol G}
1 1 1 2 # L
5 2 5 8 # U
1 0 1 2 # X
このファイルを作成したら、以下のようにband_kpoint.plを実行すればファイルkpoint.dataが作成されます。
% band_kpoint.pl bandkpt.in
kpoint.dataは以下のような記述になっています。
141 141 a.
0 50 50 100 1 b.
0 49 49 100 1
0 48 48 100 1
0 47 47 100 1
0 46 46 100 1
0 45 45 100 1
0 44 44 100 1
0 43 43 100 1
......
......
......
各項目は次のような意味です。
k点の個数を指定します。この例では141個です |
|
4つの整数は, それぞれ\(\overrightarrow{k}\)点を次式のように定義した場合の\(n_{1},n_{2},n_{3},n_{d},w\)になります(ここで \(\overrightarrow{b_1},\overrightarrow{b_2},\overrightarrow{b_3}\) は逆格子ベクトルです)。 \(\overrightarrow{k} = w \times \left( \frac{n_1}{n_d} \overrightarrow{b_1} + \frac{n_2}{n_d} \overrightarrow{b_2} + \frac{n_3}{n_d} \overrightarrow{b_3} \right)\) |
5.10.2. 固定電荷の計算を行う
バンド構造は、固定電荷計算によって計算できます。固定電荷計算とは、SCF計算によって得られた電荷密度データは「正しい」ものとして固定し、新しいバンド、k点セットで波動関数を解きなおす計算手法です。固定電荷計算については 3.3.7 章 の説明も参照してください。固定電荷計算は、SCFの計算を行ったディレクトリで実行しても問題はありませんが, 波動関数などのデータが上書きされないようにするため新たに固定電荷用の実行ディレクトリを作成することをお勧めします.
5.10.2.1. 入力パラメータ
file_names.data
file_names.dataは基本的にはSCF計算の場合と同様ですが, F_CHGT識別子でSCF計算によって得られた電荷密度ファイルを指す必要がある点が異なります. このファイルはSCF計算で利用したfile_names.data中のF_CHGT識別子で指定されるファイルであり、既定の名前はnfchgt.dataです。たとえば、SCF計算を行ったディレクトリーのすぐ下のディレクトリーにおいて固定電荷用の入力データを作成している場合、file_names.dataに以下を記述します。
&fnames
...
F_CHGT = '../nfchgt.data'
F_KPOINT = 'kpoint.data'
...
/
もしPAW法による計算を行っているのならば、F_CHGTのほかにF_CNTN_BIN_PAWという識別子によって指定されるファイルもSCF計算のファイルを指す必要があります。また、DFT+U法による 計算を行っている場合、占有行列ファイルをSCF計算のファイルをF_OCCMAT識別子によって指定する必要があります。具体的には、以下のようになります。
&fnames
...
F_CHGT = '../nfchgt.data'
F_OCCMAT = '../occmat.data' <--- DFT+Uの場合は必要
F_CNTN_BIN_PAW = '../continue_bin_paw.data' <--- PAW法の場合は必要
...
/
入力パラメータファイル
固定電荷計算用の入力ファイルを作成します。基本的にはSCF計算で利用した入力ファイルを元に作成するとよいでしょう。ただし次の点にご注意いただく必要があります
原子の座標の作成
構造緩和を行った場合固定電荷の入力ではその緩和された構造を利用する必要があります。従って、構造緩和を行った場合はF_DYNMファイルに書かれている最後の構造を参考に原子の座標を設定してください。
計算条件の変更
固定電荷で計算する、という指定を下記の要領で行います。
Control{
...
condition = fixed_charge
...
}
...
固定電荷の計算も継続計算に対応しています。継続計算を行う場合, conditionをfixed_charge_continuationとしてください。
k 点サンプリングの設定
作成したkpoint.dataを読み込むように、k点サンプリング法を以下のように編集します。
accuracy{
...
ksampling{
method = file
}
...
}
ek_convergenceブロックの設定
固有値計算の計算条件などを設定する, accuracy.ek_convergenceブロックの設定を行う必要があります。以下, ek_convergenceブロックの記述の例を示します。
accuracy{
ek_convergence{
num_max_iteration = 500
delta_eigenvalue = 1.e-5
succession = 2
num_extra_bands = 10
}
}
ek_convergenceブロックの各変数の意味は下記の通りです
num_max_iteration |
繰り返し計算の上限を指定します |
delta_eigenvalue |
収束判定を設定します。 この値のデフォルト値は1e-5です。多くの場合このデフォルト値で問題はないと思われますが、目安としては、絶縁体, 半導体の場合は1.e-4 rydberg程度, 金属の場合は1.e-6 rydberg程度がよいでしょう。 |
succession |
全エネルギーの前ステップとの差がdelta_eigenvalue以下succession回連続で収まった時点で収束したと見做します。 |
num_extra_bands |
追加するバンドの数です。 増やすことによって収束性が向上する場合があります。バンド数の一割程度が目安となります。 指定を省略すると、バンド数に応じて自動決定されます。 |
5.10.3. バンド構造図の作成
計算を実行した結果、全\(k\)点の各バンドの固有エネルギーがファイルnfenergy.dataに出力されます。
num_kpoints = 117 (a)
num_bands = 8 (b)
nspin = 1 (c)
Valence band max = 0.233846 (d)
nk_converged = 117 (e)
ik = 1 ( 0.500000 0.500000 0.000000 )
ik = 2 ( 0.487805 0.487805 0.000000 )
ik = 3 ( 0.475610 0.475610 0.000000 )
ik = 4 ( 0.463415 0.463415 0.000000 )
ik = 5 ( 0.451220 0.451220 0.000000 )
ik = 6 ( 0.439024 0.439024 0.000000 )
...
...
...
=== energy_eigen_values ===
ik = 1 ( 0.000000 0.500000 0.500000 ) (f)
-0.0484324576 -0.0484324576 0.1258094928 0.1258094928 (g)
0.2619554301 0.2619554301 0.6015285208 0.6015285208
=== energy_eigen_values ===
ik = 2 ( 0.000000 0.490000 0.490000 )
-0.0540717201 -0.0427149632 0.1258687739 0.1258687739
0.2607026807 0.2633829927 0.6006243932 0.6006243932
......
......
......
各項目を説明します。
k 点数。この例では141 個です。 |
|
バンド数。この例では8 です。 |
|
スピン自由度。1 か2 の値をとります。この例では1 であり、スピン分極を考慮しない計算に対応します。 |
|
フェルミエネルギーの値。半導体/絶縁体の場合価電子帯の上端のエネルギーが記述されます。単位はハートリーです。 |
|
計算したk 点 |
|
固有値の情報が記述されます。まずこの行で, どのk点に対応する固有値データかが分かります。この例では, 1 番目のkで, その座標は逆格子ベクトルを基底として(0,0.5,0.5)となります。 |
|
固有値のデータがバンドの数だけ出力されます。単位はハートリーです。 |
スピンを考慮した計算の場合(上記の(c) が2 の場合) もほぼ同様のファイル形式ですが, 上記の(e) の隣に“UP”か“DOWN” と記述される, という違いがあります。それぞれ多数派スピンと少数派スピンに対応する固有値が書き出されます。
......
......
......
=== energy_eigen_values ===
ik = 1 ( 0.000000 0.000000 0.000000) UP
-0.1998699758 0.0267639589 0.0267639589 0.0267639589
0.0725171077 0.0725171077 1.0289118953 1.0289118953
1.0289118953 1.1650173104 1.1650173104 1.1650173104
1.2129026022 1.2129026022 1.2994754011 1.2994754011
1.2994754011 1.6365336765 2.2629596795 2.2629596795
=== energy_eigen_values ===
ik = 2 ( 0.000000 0.000000 0.000000) DOWN
-0.1960420390 0.1062941746 0.1062941746 0.1062941746
0.1799862148 0.1799862148 1.0183970612 1.0183970612
1.0183970612 1.2174266166 1.2174266166 1.2192701193
1.2192701193 1.2192701193 1.3289165100 1.3289165100
1.3289165100 1.6910264603 2.2876818717 2.2876818717
......
......
......
このようなデータからバンド構造図を作成するのは手間がかかりますが、PHASEにはこの結果からバンド構造図を簡単に作成する“band.pl”というPerlスクリプトが付属しています。 band.plは、以下のように実行します。
% band.pl nfenergy.data bandkpt.in -erange=-10,10 -color -with_fermi
例として、体心立方鉄のバンド構造図を 図 5.14 に示します。

図 5.14 体心立方鉄のバンド構造図。
5.11. 格子定数
5.11.1. ストレステンソルを利用したユニットセル最適化機能
PHASE/0には、ストレステンソルを利用して単位胞を最適化する機能が備わっています。ここでは、この機能の使い方の説明を行います。
5.11.1.1. 入力パラメータ
まずは、通常通り入力パラメータファイルを記述します。セルを変形させたあとに座標の緩和を行いたい場合は通常通り原子座標最適化用のパラメータを設定すればセルの変形→力が収束していない場合は原子座標の最適化、という動作をするようになります。さらに、単位胞最適化用の、以下のような設定を加えます。
structure_evolution{
lattice{
sw_optimize_lattice = on
}
}
変数sw_optimize_latticeをonとすると本機能を利用することができます。latticeブロックには、以下の変数を定義することが可能です。
sw_optimize_lattice |
単位胞最適化機能を有効にする場合onとします。 デフォルト値はoffです。 なお、このスイッチがonの場合はsw_stressは自動的にonになります。 |
sw_uniform |
単位胞を一様に変化させたい場合にonとします。デフォルト値はoffです。 このパラメータがonの場合、ストレステンソルの対角要素の平均値によって体積を変化させるように動作します。 |
sw_rebuild_pws |
単位胞を変形させた後に平面波基底を作り直すかどうかを指定します。 デフォルト値はon, つまり格子が変形する度に平面波を作り直します。offとすることによって電子状態計算の収束性を向上させることができますが、格子が変形しても同じ平面波セットを利用している、ということは厳密にはカットオフエネルギーが微妙に変化している、ということに相当する点に注意が必要です。また、このパラメータをoffとすると継続計算ができなくなってしまいます。 |
method |
最適化の手法を選択します。 bfgs, quench, steepest_descent のいずれかを指定します。 デフォルト値はbfgsです。 |
delta_stress |
methodがquenchかsteepest_descent の場合の更新の刻み幅を指定します。 デフォルト値は1です。 |
max_stress |
収束判定に利用する、ストレステンソルの最大値を圧力の単位で指定します。 デフォルト値は 1.e-6 hartree/bohr3 です。 sw_uniformがonの場合は ストレステンソルの対角要素の平均が収束判定に採用されます。 |
sw_optimize_coordinates_once |
原子配置の最適化は1回目の格子の更新時のみ行いたい場合にonとします。 |
fix_length_a |
onとするとa軸の長さを固定して格子を最適化します。 |
fix_length_b |
onとするとb軸の長さを固定して格子を最適化します。 |
fix_length_c |
onとするとc軸の長さを固定して格子を最適化します。 |
fix_angle_alpha |
onとすると格子定数αを固定して格子を最適化します。 |
fix_angle_beta |
onとすると格子定数βを固定して格子を最適化します。 |
fix_angle_gamma |
onとすると格子定数γを固定して格子を最適化します。 |
5.11.1.2. ストレステンソルの補正
PHASE/0によるストレステンソルの計算は、精度が低い場合があります。原因は、“格子がひずむことによる平面波数の変化の効果”がとりいれられていないからです。この効果を取り入れることによって、ある程度補正を行うことが可能です。
方法1.
運動エネルギーの計算におけるGベクトルの高周波成分をスメアすることによって、“平面波数一定”の状況を“カットオフエネルギー一定”の状況に近づけることができます。 [Bernasconi95] では、運動エネルギーの高周波成分を以下のように置き換えることが提案されています。
PHASE/0では、上式を利用したストレステンソルの計算を行うことができます。以下のような設定を入力パラメーターファイルに記述します。
structure_evolution{
lattice{ sw_optimize_lattice = on }
stress{
sw_smear_KE = on
a = 15 rydberg
sigma = 0.1 rydberg
e0 = 35 rydberg
}
}
structure_evolutionの下にstressブロックを作成し、設定を行います。sw_smear_KE=onとするとこの機能が有効になります。a, sigma, e0には対応するパラメーターを指定します。
デフォルト値はa=0.375, ecut, sigma = 0.1 Rydberg, e0=ecut-1 Rydbergです。
方法2.(バージョン2019.01以降)
複数のカットオフエネルギーによる計算から誤差を見積もることができます。ターゲットカットオフエネルギーを \(E_{\mathrm c}\) 、変化量を \(\Delta E_{\mathrm c}\) 全エネルギーの変化量を \(\Delta E_{\mathrm t}\) とすると、ストレスの誤差 \(\sigma_{\mathrm e}\) は以下のように見積もることができます。
この補正をPHASE/0に計算させるには、以下のようにstressブロックにおいてsw_stress_correctionをonとします。
structure_evolution{
stress{
sw_stress = on
sw_stress_correction = on
}
}
ストレステンソルの補正は、カットオフエネルギーを変化させてストレステンソルを求めることによって計算します。どの程度カットオフエネルギーを変化させるかはdelta_ecutによって指定します。カットオフエネルギーをecut-delta_ecutとしたケースとecut+delta_ecutとしたケースのストレステンソル計算が行われ、その後補正が計算されます。なお、補正の計算前に計算が終了した場合継続できないので注意が必要です。補正値は以下のようにoutput000ファイルに記録されます。
!** Pulay stress : -0.000194696412156
補正が計算されたあと、入力パラメーターファイルに格子最適化の設定が行われている場合補正を組み込んだ状態で格子最適化計算が始まります。そうでない場合、以下の要領で補正の値を入力ファイルに書き込み、格子最適化などを行う設定にしたうえで再度計算を実行してください(補正が必要なのは対角要素のみ、また誤差が-0.0001auだったとして)。
structure_evolution{
lattice{
sw_optimize_lattice = on
external_stress{
s11 = -0.0001
s22 = -0.0001
s33 = -0.0001
}
}
}
検証
これらの補正を利用し、TiO2の格子定数を計算した結果を以下の表にまとめました。方法1.のパラメーターはデフォルト値、方法2.のは±5 Rydbergとしました。
a (bohr) |
c (bohr) |
|
|---|---|---|
カットオフ36 Rydberg, EV曲線 |
8.8017 |
5.6355 |
カットオフ36 Rydberg, 補正なし |
8.6825 |
5.5862 |
カットオフ36 Rydberg, 方法 1. |
8.7593 |
5.6072 |
カットオフ36 Rydberg, 方法 2. |
8.8052 |
5.6200 |
カットオフ80 Rydberg, 補正なし |
8.7918 |
5.6158 |
方法1.2.とも改善しています。特に、方法2.を使うとEV曲線からもとめた格子定数とほぼ同じ格子定数が得られています。
M. Bernasconi, G.L.Chiarotti, P.Focher,S.Scandolo,E.Tosatti,M.Parrinello Journal of Physics and Chemistry of Solids, 56 501-505 (1995).
5.11.1.3. 格子と原子座標を同時に最適化する方法(バージョン2023.01以降)
ストレステンソルを用いた格子の最適化においては、まずは原子座標の最適化が行われ、収束後格子の最適化が行われます。 この二種類の最適化を同時に進行させることができます。 この機能を利用するためには、入力パラメーターファイルに以下の記述を行います。
structure_evolution {
lattice{
sw_optimize_coords_sametime = on
}
}
多くの場合このオプションを使うと少ないステップ数で最終的な収束を得ることができます。
本機能を利用する場合は、格子緩和手法はBFGS(既定値)のみ使用できます。 また、sw_uniform=OFF(既定値)でご利用ください。
5.11.1.4. 計算結果の出力
結果はoutput000ファイル、nfefn.dataファイル、nfdynm.dataファイルに記録されます。
output000ファイルには、ストレステンソルが記録されます。以下のコマンドによってその情報を抽出することができます。
% grep –A3 ‘Total STRESS’ output000
Total STRESS TENSOR
0.0002320404 0.0000000000 0.0000000000
0.0000000000 0.0002320404 -0.0000000000
0.0000000000 -0.0000000000 0.0002117384
--
Total STRESS TENSOR
0.0002266097 -0.0000000000 -0.0000000000
-0.0000000000 0.0002266097 0.0000000000
-0.0000000000 0.0000000000 0.0002051476
--
...
...
通常の計算の場合ストレステンソルが1組出力されるのみですが、本機能を利用している場合はストレステンソルの履歴が出力されます。
nfefn.dataファイルには、通常通り全エネルギーや原子に働く力の最大値のほか、ストレステンソルの最大値(sw_uniformがonの場合は対角要素の平均値)が記録されます。たとえば、以下のような出力が得られます。
iter_unitcell, iter_ion, iter_total, etotal, forcmx, stressmx
1 1 18 -181.4043211413 0.0020128619
1 2 27 -181.4043355689 0.0015666906
1 3 36 -181.4043464493 0.0011267018
1 4 44 -181.4043509953 0.0008837770
1 5 53 -181.4043582176 0.0000137026 0.0002326236
2 1 73 -181.4044226903 0.0000645338 0.0002272841
nfdynm.dataファイルも通常のものとほぼ同じですが、通常の計算の場合は一度しか出力されないヘッダーが、セルベクトルが変形される度に出力されます。
#
# a_vector = 8.6795114819 0.0000000000 0.0000000000
# b_vector = 0.0000000000 8.6795114819 0.0000000000
# c_vector = 0.0000000000 0.0000000000 5.5916992108
# ntyp = 2 natm = 6
# (natm->type) 2 2 1 1 1 1
# (speciesname) 1 : O
# (speciesname) 2 : Ti
#
cps and forc at (iter_ion, iter_total = 1 18 )
1 0.000000000 0.000000000 0.000000000 0.000000 0.000000 0.000000
2 4.339755741 4.339755741 2.795849605 0.000000 0.000000 0.000000
3 2.643779197 2.643779197 0.000000000 -0.001423 -0.001423 0.000000
4 6.983534938 1.695976544 2.795849605 -0.001423 0.001423 0.000000
……
……
#
# a_vector = 8.7672856463 0.0000000000 0.0000000000
# b_vector = 0.0000000000 8.7672856463 0.0000000000
# c_vector = 0.0000000000 0.0000000000 5.6429940606
# ntyp = 2 natm = 6
# (natm->type) 2 2 1 1 1 1
# (speciesname) 1 : O
# (speciesname) 2 : T
#
cps and forc at (iter_ion, iter_total = 1 111 )
1 0.000000000 0.000000000 0.000000000 0.000000 0.000000 0.000000
2 4.383642823 4.383642823 2.821497030 0.000000 0.000000 0.000000
3 2.663907294 2.663907294 0.000000000 0.001773 0.001773 0.000000
4 7.047550117 1.719735530 2.821497030 0.001773 -0.001773 0.000000
5 1.719735530 7.047550117 2.821497030 -0.001773 0.001773 0.000000
6 -2.663907294 -2.663907294 0.000000000 -0.001773 -0.001773 0.000000
……
……
5.11.1.5. 計算例:ルチル型TiO2 (ストレス補正なし)
ルチル型TiO 2 の格子最適化を行った例を紹介します。入力データは samples/unitcel_optimization/TiO2 以下にあります。
入力パラメータファイルには、以下のような設定を施しました。
カットオフエネルギーは80 Rydberg
擬ポテンシャルはポータルサイトにおいて公開されているTi_ggapbe_paw_us_02.ppとO_ggapbe_paw_us_02m.pp
原子座標の最適化を施す設定;手法はBFGS法、収束判定となる力の最大値は2e-4
初期原子配置および格子定数は、無機材料データベースAtomWork(http://crystdb.nims.go.jp/)に登録されていたルチル型TiO2のデータを採用
波動関数ソルバー、電荷密度ミキサーは指定せず、デフォルト設定を採用。
採用したカットオフエネルギーは80 Rydbergと比較的大きなものですが、後述のようにTiO2の場合はこれくらい必要であると考えられます。
nfefn.dataファイルの内容は、以下のようになりました。
iter_unitcell, iter_ion, iter_total, etotal, forcmx, stressmx
1 1 18 -181.4043211413 0.0020128619
1 2 27 -181.4043355689 0.0015666906
1 3 36 -181.4043464493 0.0011267018
1 4 44 -181.4043509953 0.0008837770
1 5 53 -181.4043582176 0.0000137026 0.0002326236
2 1 73 -181.4044226903 0.0000645338 0.0002272841
3 1 92 -181.4044839579 0.0001241955 0.0002222588
4 1 111 -181.4056948858 0.0025074070 0.0002222588
4 2 120 -181.4057176163 0.0020195652 0.0002222588
4 3 130 -181.4057600852 0.0000156213 0.0000444895
……
……
9 1 248 -181.4058191217 0.0001647915 0.0000332105
10 1 268 -181.4058328662 0.0000709369 0.0000119789
11 1 287 -181.4058349707 0.0000268520 0.0000015502
12 1 306 -181.4058351835 0.0000244918 0.0000006790
まずは、原子座標の最適化が5回実施されています。その間ストレステンソルは未計算なので、6列目は空欄になっています。5回目で原子に働く力の最大値が閾値より小さくなったので、セルを変形させたのちに計算が進行しています。この際に、単位胞最適化の更新回数を表す1列目の数値が2になっていることがわかります。また、6列目にストレステンソルの最大値が記録されています。2回目と3回目の更新時はセルを変形させても原子に働く力の最大値は閾値以下だったので原子座標の最適化は実施されませんでしたが、4回目セルベクトル更新時にはそうではなかったので原子座標の最適化が行われています。このようにセルの最適化と必要に応じた原子座標の最適化が行われつつ計算が進行し、セルの更新回数が12回となったところでストレステンソルの最大値が閾値以下となったので計算は収束したとみなされ終了しています。単位胞最適化収束の履歴を、図にまとめました。

図 5.15 単位胞最適化の履歴。赤線:全エネルギー、緑線:ストレステンソルの最大成分。
安定な格子定数は、nfdynm.dataファイルに記録された最後のセルベクトル更新の情報からもとめることができます。この例の場合、a=8.7934 bohr, c=5.6164 bohrと得られました。
ストレステンソルとカットオフエネルギー
ストレステンソルは、全エネルギーや原子間力と比較してカットオフエネルギーに対して収束しづらい傾向があります。例として、ルチル型TiO2の、実測値の格子定数で計算したストレステンソルとカットオフエネルギーの関係を図にプロットしました。
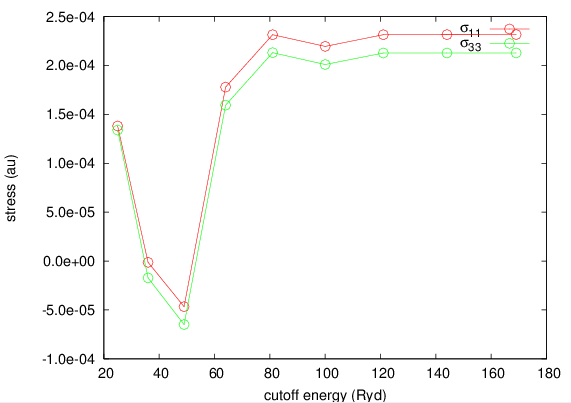
図 5.16 ルチル型TiO2の場合の、ストレステンソルとカットオフエネルギーの関係
図からわかるように、カットオフ50 Rydberg程度の場合ストレステンソルの符号が間違ってしまっています。このケースでは、ある程度収束したストレステンソルを得るためには、最低でも80 Rydberg程度以上のカットオフエネルギーが必要であることが示唆されます。
5.11.1.6. 計算例:ルチル型TiO2 (ストレス補正あり、バージョン2019.01以降)
5.11.1.5 章 で行った最適化を、ストレス補正を有効にして実行してみます。
入力データは samples/unitcell_optimization/TiO2_with_correction にあります。
入力パラメータファイルには、以下のような設定を施しました。 計算条件は下記の通り。
カットオフエネルギーは36 Rydberg
ストレス補正は有効 (structure_evolutionブロックの下のstressブロックにおいてsw_stress_correction = onと設定)
擬ポテンシャルはポータルサイトにおいて公開されているTi_ggapbe_paw_us_02.ppとO_ggapbe_paw_us_02m.pp
原子座標の最適化を施す設定;手法はBFGS法、収束判定となる力の最大値は2e-4
初期原子配置および格子定数は、無機材料データベースAtomWork( http://crystdb.nims.go.jp/ )に登録されていたルチル型TiO2のデータを採用
波動関数ソルバー、電荷密度ミキサーは指定せず、デフォルト設定を採用。
結果得られる単位胞最適化収束の履歴を次の図に示します。比較的低いカットオフ(36 Rydberg)を採用しているにも関わらずスムーズな最適化が行えました。結果得られる格子定数は、a = 8.806 bohr, c = 5.619 bohrとなりました。

図 5.17 単位胞最適化の履歴。赤線:全エネルギー、緑線:ストレステンソルの最大成分。
5.11.1.7. 計算例:ルチル型TiO2 (ストレス補正あり、原子座標と格子同時最適化、バージョン2023.01以降)
5.11.1.5 章 で行った最適化を、ストレス補正を有効にし、さらに原子座標と格子を同時に最適化します。
入力データは samples/unitcell_optimization/TiO2_with_correction_sametime にあります。
入力設定は sw_optimize_coords_sametime=on とした以外は 5.11.1.6 章 の場合と同一です。
手法 |
総SCF計算回数 |
格子定数 \(a\) (Bohr) |
格子定数 \(c\) (Bohr) |
全エネルギー(Hartree) |
sw_optimize_coords_sametime=off |
208 |
8.806 |
5.619 |
-181.3054220253 |
sw_optimize_coords_sametime=on |
125 |
8.806 |
5.620 |
-181.3054294087 |
両手法でほぼ同じ結果を得ることができましたが、 sw_optimize_coords_sametime=on とするとSCFの回数を4割程度削減することができました。この問題に限らず、多くの問題で計算時間の短縮が見込めます。
5.11.2. マーナハンの状態方程式を用いた格子定数の計算
格子定数は、複数の格子定数において全エネルギーを計算することによって計算することが可能です。 特に、立方晶の場合は次のマーナハンの状態方程式にフィットすることによって格子定数だけではなく体積弾性率も求めることが可能です。
ここで\(E_{\text{tot}}\left( V \right)\)は単位胞の体積\(V\)における全エネルギー、\(B\)は体積弾性率、\(B^{'}\)は体積弾性率の体積微分、\(V_{0}\)は安定な格子定数における単位胞の体積です。 \(B,B^{'},V_{0},E_{\text{tot}}\left( V_{0} \right)\)の4つがフィッティングパラメータです。
5.11.2.1. 計算例:Si 結晶
Si結晶の格子定数の計算例です。この例題は、 samples/unitcell_optimization/murnaghan_Si です。
このディレクトリーの下には、さらにvolxxxというサブディレクトリが存在します。各々のサブディレクトリは、xxxという単位胞の体積に対応した入力データが格納されています。
たとえば、vol1200というディレクトリにおける計算モデルの指定は以下のようになっています。
structure{
element_list{
#tag element atomicnumber
Si 14
}
atom_list{
atoms{
#units angstrom
#tag element rx ry rz
Si 0.125 0.125 0.125
Si -0.125 -0.125 -0.125
}
coordinate_system = internal
}
unit_cell{
a_vector = 10.62658569182611066038 0 0
b_vector = 0 10.62658569182611066038 0
c_vector = 0 0 10.62658569182611066038
}
symmetry{
method = automatic
tspace{
lattice_system = facecentered
}
sw_inversion = on
}
unit_cell_type = bravais
}
座標データは、フラクショナルな座標データで指定しています。 カルテシアンでもよいのですが、格子定数を変えるたびに座標値も変えるのは手間がかかるので、格子定数の計算にはフラクショナル座標が適していると言えます。
unit_cell_typeとしてbravaisを採用し、さらにtspaceの下のlattice_systemにfacecenteredを指定しています。 このようにすることによって、格子定数の指定がしやすいブラベー格子によって入力の格子を指定し、実際の計算はより負荷の少ない基本格子で行うことが可能となります。
実際の計算はブラベー格子ではなく基本格子で行われるので、体積としてブラベー格子の値を採用するのであれば必要に応じて結果を変換する必要があります。 たとえば、この例の場合体積弾性率は得られる値の4倍にします(面心立方格子のブラベー格子の体積は基本格子の4倍のため)。
計算を行い、マーナハンの状態方程式にフィットした結果を 図 5.18 および 表 5.7 に示します。別途計算した原子の全エネルギーから、凝集エネルギーも示しています。 原子あたりの凝集エネルギーは、原子の全エネルギーから最安定の格子定数における結晶の全エネルギーを原子数で割った値を引くことによって得ることができます。

図 5.18 シリコンのEnergy-Volume曲線。白丸は計算値、実線はフィットした結果。
PHASE |
実測データ |
|
|---|---|---|
a (Å) |
5.48 |
5.43 |
B (GPa) |
87.5 |
98.8 |
Ecoh (eV/atom) |
4.60 |
4.63 |